 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Systematic Replicability and Comparative Study of BSARec and SASRec for Sequential Recommendation
Jun 17, 2025This study aims at comparing two sequential recommender systems: Self-Attention based Sequential Recommendation (SASRec), and Beyond Self-Attention based Sequential Recommendation (BSARec) in order to check the improvement frequency enhancement - the added element in BSARec - has on recommendations. The models in the study, have been re-implemented with a common base-structure from EasyRec, with the aim of obtaining a fair and reproducible comparison. The results obtained displayed how BSARec, by including bias terms for frequency enhancement, does indeed outperform SASRec, although the increases in performance obtained, are not as high as those presented by the authors. This work aims at offering an overview on existing methods, and most importantly at underlying the importance of implementation details for performance comparison.
A Theoretical Analysis of Recommendation Loss Functions under Negative Sampling
Nov 12, 2024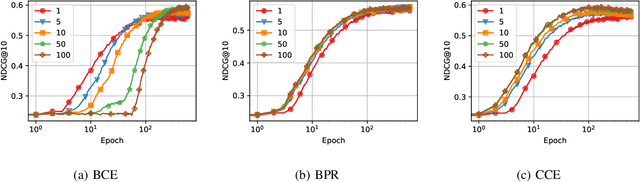
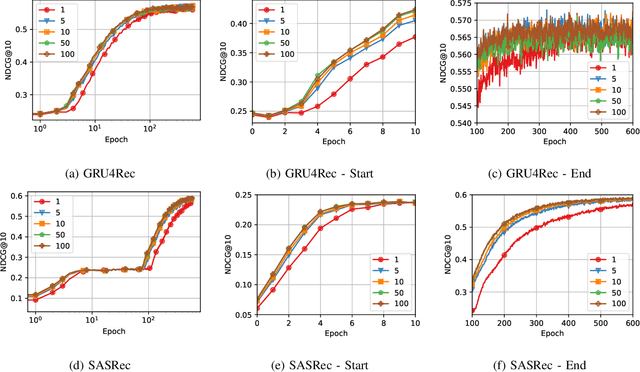
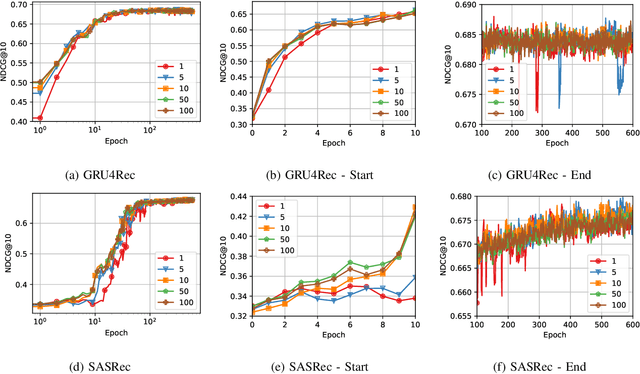
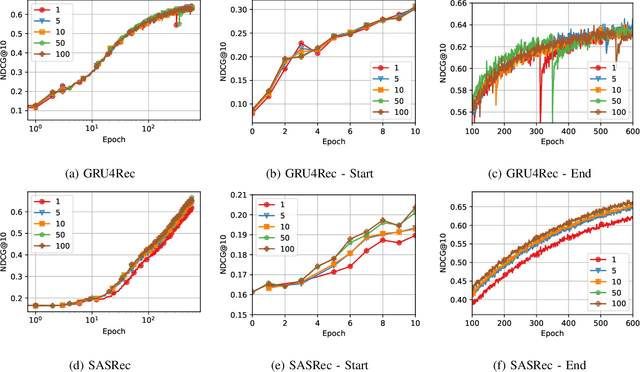
Recommender Systems (RSs) are pivotal in diverse domains such as e-commerce, music streaming, and social media. This paper conducts a comparative analysis of prevalent loss functions in RSs: Binary Cross-Entropy (BCE), Categorical Cross-Entropy (CCE), and Bayesian Personalized Ranking (BPR). Exploring the behaviour of these loss functions across varying negative sampling settings, we reveal that BPR and CCE are equivalent when one negative sample is used. Additionally, we demonstrate that all losses share a common global minimum. Evaluation of RSs mainly relies on ranking metrics known as Normalized Discounted Cumulative Gain (NDCG) and Mean Reciprocal Rank (MRR). We produce bounds of the different losses for negative sampling settings to establish a probabilistic lower bound for NDCG. We show that the BPR bound on NDCG is weaker than that of BCE, contradicting the common assumption that BPR is superior to BCE in RSs training. Experiments on five datasets and four models empirically support these theoretical findings. Our code is available at \url{https://anonymous.4open.science/r/recsys_losses} .
A graph neural network-based model with Out-of-Distribution Robustness for enhancing Antiretroviral Therapy Outcome Prediction for HIV-1
Dec 29, 2023


Predicting the outcome of antiretroviral therapies for HIV-1 is a pressing clinical challenge, especially when the treatment regimen includes drugs for which limited effectiveness data is available. This scarcity of data can arise either due to the introduction of a new drug to the market or due to limited use in clinical settings. To tackle this issue, we introduce a novel joint fusion model, which combines features from a Fully Connected (FC) Neural Network and a Graph Neural Network (GNN). The FC network employs tabular data with a feature vector made up of viral mutations identified in the most recent genotypic resistance test, along with the drugs used in therapy. Conversely, the GNN leverages knowledge derived from Stanford drug-resistance mutation tables, which serve as benchmark references for deducing in-vivo treatment efficacy based on the viral genetic sequence, to build informative graphs. We evaluated these models' robustness against Out-of-Distribution drugs in the test set, with a specific focus on the GNN's role in handling such scenarios. Our comprehensive analysis demonstrates that the proposed model consistently outperforms the FC model, especially when considering Out-of-Distribution drugs. These results underscore the advantage of integrating Stanford scores in the model, thereby enhancing its generalizability and robustness, but also extending its utility in real-world applications with limited data availability. This research highlights the potential of our approach to inform antiretroviral therapy outcome prediction and contribute to more informed clinical decisions.
Incorporating temporal dynamics of mutations to enhance the prediction capability of antiretroviral therapy's outcome for HIV-1
Nov 08, 2023Motivation: In predicting HIV therapy outcomes, a critical clinical question is whether using historical information can enhance predictive capabilities compared with current or latest available data analysis. This study analyses whether historical knowledge, which includes viral mutations detected in all genotypic tests before therapy, their temporal occurrence, and concomitant viral load measurements, can bring improvements. We introduce a method to weigh mutations, considering the previously enumerated factors and the reference mutation-drug Stanford resistance tables. We compare a model encompassing history (H) with one not using it (NH). Results: The H-model demonstrates superior discriminative ability, with a higher ROC-AUC score (76.34%) than the NH-model (74.98%). Significant Wilcoxon test results confirm that incorporating historical information improves consistently predictive accuracy for treatment outcomes. The better performance of the H-model might be attributed to its consideration of latent HIV reservoirs, probably obtained when leveraging historical information. The findings emphasize the importance of temporal dynamics in mutations, offering insights into HIV infection complexities. However, our result also shows that prediction accuracy remains relatively high even when no historical information is available. Supplementary information: Supplementary material is available.
Unboxing Tree Ensembles for interpretability: a hierarchical visualization tool and a multivariate optimal re-built tree
Feb 15, 2023The interpretability of models has become a crucial issue in Machine Learning because of algorithmic decisions' growing impact on real-world applications. Tree ensemble methods, such as Random Forests or XgBoost, are powerful learning tools for classification tasks. However, while combining multiple trees may provide higher prediction quality than a single one, it sacrifices the interpretability property resulting in "black-box" models. In light of this, we aim to develop an interpretable representation of a tree-ensemble model that can provide valuable insights into its behavior. First, given a target tree-ensemble model, we develop a hierarchical visualization tool based on a heatmap representation of the forest's feature use, considering the frequency of a feature and the level at which it is selected as an indicator of importance. Next, we propose a mixed-integer linear programming (MILP) formulation for constructing a single optimal multivariate tree that accurately mimics the target model predictions. The goal is to provide an interpretable surrogate model based on oblique hyperplane splits, which uses only the most relevant features according to the defined forest's importance indicators. The MILP model includes a penalty on feature selection based on their frequency in the forest to further induce sparsity of the splits. The natural formulation has been strengthened to improve the computational performance of mixed-integer software. Computational experience is carried out on benchmark datasets from the UCI repository using a state-of-the-art off-the-shelf solver. Results show that the proposed model is effective in yielding a shallow interpretable tree approximating the tree-ensemble decision function.
AI-based Data Preparation and Data Analytics in Healthcare: The Case of Diabetes
Jun 13, 2022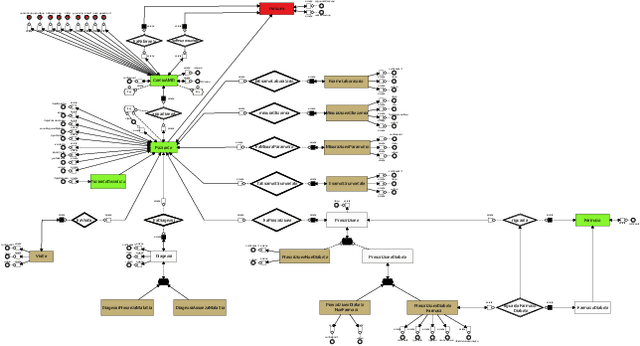
The Associazione Medici Diabetologi (AMD) collects and manages one of the largest worldwide-available collections of diabetic patient records, also known as the AMD database. This paper presents the initial results of an ongoing project whose focus is the application of Artificial Intelligence and Machine Learning techniques for conceptualizing, cleaning, and analyzing such an important and valuable dataset, with the goal of providing predictive insights to better support diabetologists in their diagnostic and therapeutic choices.