 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCoNIC Challenge: Pushing the Frontiers of Nuclear Detection, Segmentation, Classification and Counting
Mar 14, 2023



Nuclear detection, segmentation and morphometric profiling are essential in helping us further understand the relationship between histology and patient outcome. To drive innovation in this area, we setup a community-wide challenge using the largest available dataset of its kind to assess nuclear segmentation and cellular composition. Our challenge, named CoNIC, stimulated the development of reproducible algorithms for cellular recognition with real-time result inspection on public leaderboards. We conducted an extensive post-challenge analysis based on the top-performing models using 1,658 whole-slide images of colon tissue. With around 700 million detected nuclei per model, associated features were used for dysplasia grading and survival analysis, where we demonstrated that the challenge's improvement over the previous state-of-the-art led to significant boosts in downstream performance. Our findings also suggest that eosinophils and neutrophils play an important role in the tumour microevironment. We release challenge models and WSI-level results to foster the development of further methods for biomarker discovery.
Simultaneous Semantic and Instance Segmentation for Colon Nuclei Identification and Counting
Mar 01, 2022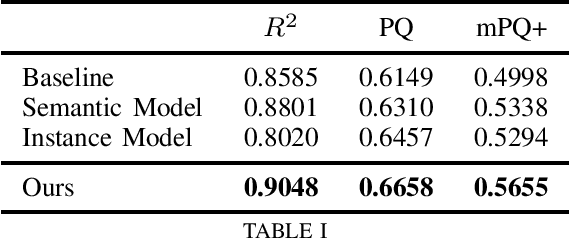
We address the problem of automated nuclear segmentation, classification, and quantification from Haematoxylin and Eosin stained histology images, which is of great relevance for several downstream computational pathology applications. In this work, we present a solution framed as a simultaneous semantic and instance segmentation framework. Our solution is part of the Colon Nuclei Identification and Counting (CoNIC) Challenge. We first train a semantic and instance segmentation model separately. Our framework uses as backbone HoverNet and Cascade Mask-RCNN models. We then ensemble the results with a custom Non-Maximum Suppression embedding (NMS). In our framework, the semantic model computes a class prediction for the cells whilst the instance model provides a refined segmentation. We demonstrate, through our experimental results, that our model outperforms the provided baselines by a large margin.
Multi-modal Self-supervised Pre-training for Regulatory Genome Across Cell Types
Nov 03, 2021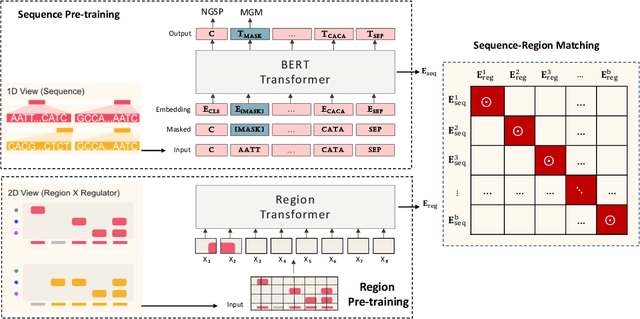

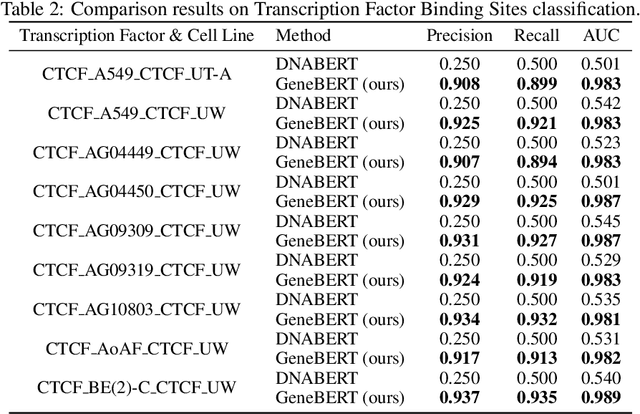
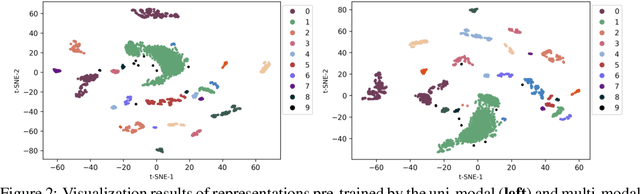
In the genome biology research, regulatory genome modeling is an important topic for many regulatory downstream tasks, such as promoter classification, transaction factor binding sites prediction. The core problem is to model how regulatory elements interact with each other and its variability across different cell types. However, current deep learning methods often focus on modeling genome sequences of a fixed set of cell types and do not account for the interaction between multiple regulatory elements, making them only perform well on the cell types in the training set and lack the generalizability required in biological applications. In this work, we propose a simple yet effective approach for pre-training genome data in a multi-modal and self-supervised manner, which we call GeneBERT. Specifically, we simultaneously take the 1d sequence of genome data and a 2d matrix of (transcription factors x regions) as the input, where three pre-training tasks are proposed to improve the robustness and generalizability of our model. We pre-train our model on the ATAC-seq dataset with 17 million genome sequences. We evaluate our GeneBERT on regulatory downstream tasks across different cell types, including promoter classification, transaction factor binding sites prediction, disease risk estimation, and splicing sites prediction. Extensive experiments demonstrate the effectiveness of multi-modal and self-supervised pre-training for large-scale regulatory genomics data.