 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRecombination of Artificial Neural Networks
Jan 12, 2019
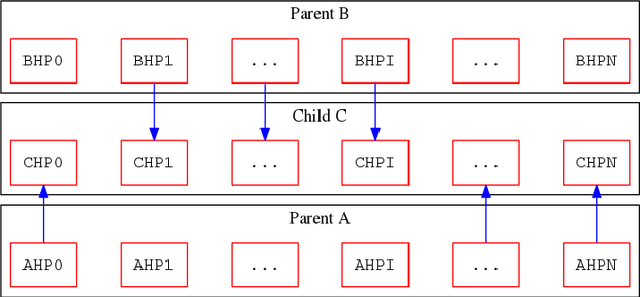

We propose a genetic algorithm (GA) for hyperparameter optimization of artificial neural networks which includes chromosomal crossover as well as a decoupling of parameters (i.e., weights and biases) from hyperparameters (e.g., learning rate, weight decay, and dropout) during sexual reproduction. Children are produced from three parents; two contributing hyperparameters and one contributing the parameters. Our version of population-based training (PBT) combines traditional gradient-based approaches such as stochastic gradient descent (SGD) with our GA to optimize both parameters and hyperparameters across SGD epochs. Our improvements over traditional PBT provide an increased speed of adaptation and a greater ability to shed deleterious genes from the population. Our methods improve final accuracy as well as time to fixed accuracy on a wide range of deep neural network architectures including convolutional neural networks, recurrent neural networks, dense neural networks, and capsule networks.
Multimodal Deep Neural Networks using Both Engineered and Learned Representations for Biodegradability Prediction
Sep 13, 2018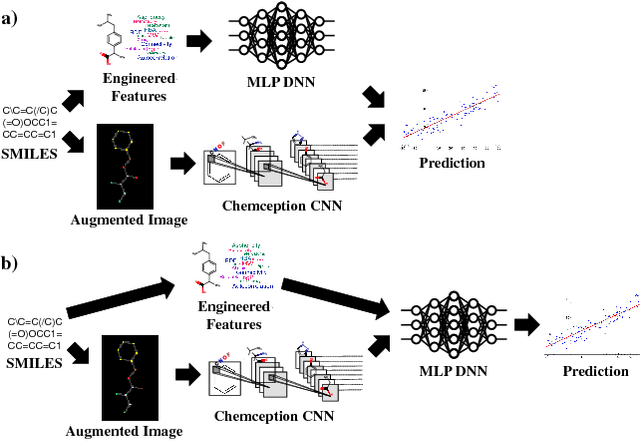

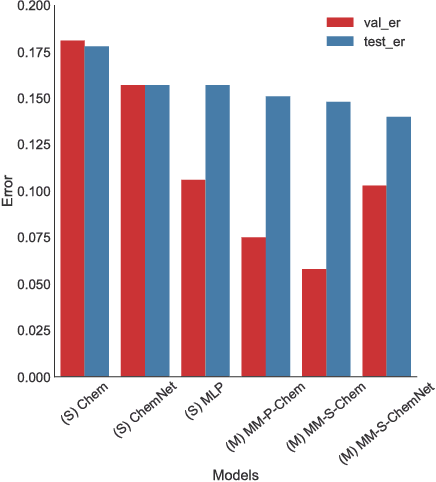
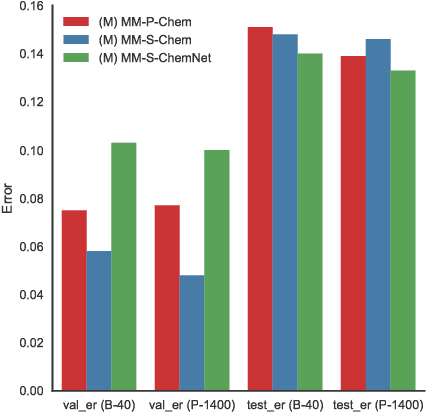
Deep learning algorithms excel at extracting patterns from raw data, and with large datasets, they have been very successful in computer vision and natural language applications. However, in other domains, large datasets on which to learn representations from may not exist. In this work, we develop a novel multimodal CNN-MLP neural network architecture that utilizes both domain-specific feature engineering as well as learned representations from raw data. We illustrate the effectiveness of such network designs in the chemical sciences, for predicting biodegradability. DeepBioD, a multimodal CNN-MLP network is more accurate than either standalone network designs, and achieves an error classification rate of 0.125 that is 27% lower than the current state-of-the-art. Thus, our work indicates that combining traditional feature engineering with representation learning can be effective, particularly in situations where labeled data is limited.
ColdRoute: Effective Routing of Cold Questions in Stack Exchange Sites
Jul 02, 2018
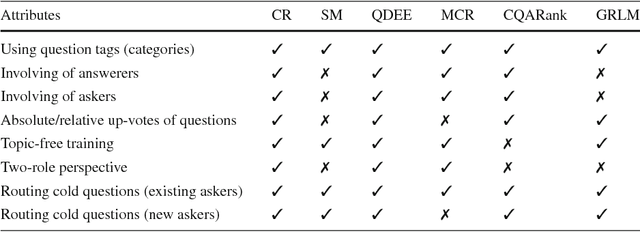
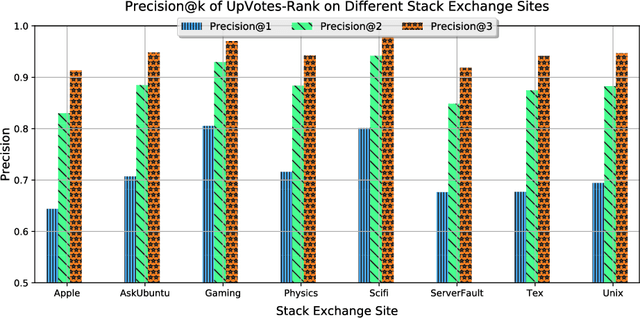
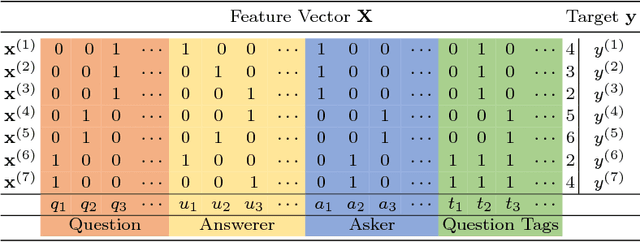
Routing questions in Community Question Answer services (CQAs) such as Stack Exchange sites is a well-studied problem. Yet, cold-start -- a phenomena observed when a new question is posted is not well addressed by existing approaches. Additionally, cold questions posted by new askers present significant challenges to state-of-the-art approaches. We propose ColdRoute to address these challenges. ColdRoute is able to handle the task of routing cold questions posted by new or existing askers to matching experts. Specifically, we use Factorization Machines on the one-hot encoding of critical features such as question tags and compare our approach to well-studied techniques such as CQARank and semantic matching (LDA, BoW, and Doc2Vec). Using data from eight stack exchange sites, we are able to improve upon the routing metrics (Precision$@1$, Accuracy, MRR) over the state-of-the-art models such as semantic matching by $159.5\%$,$31.84\%$, and $40.36\%$ for cold questions posted by existing askers, and $123.1\%$, $27.03\%$, and $34.81\%$ for cold questions posted by new askers respectively.
* Accepted to the Journal Track of The European Conference on Machine Learning and Principles and Practice of Knowledge Discovery in Databases (ECML PKDD 2018); Published by Springer: https://link.springer.com/article/10.1007%2Fs10618-018-0577-7
How Much Chemistry Does a Deep Neural Network Need to Know to Make Accurate Predictions?
Mar 18, 2018
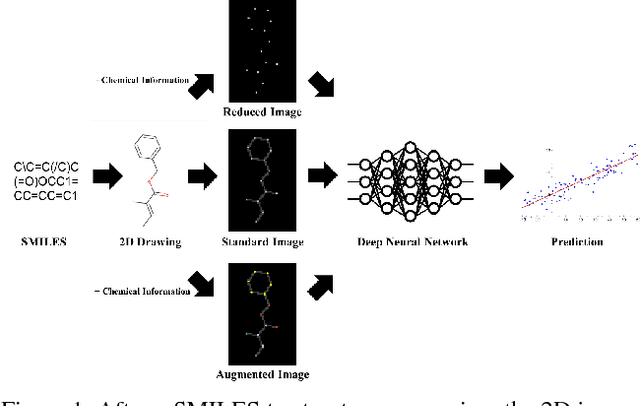
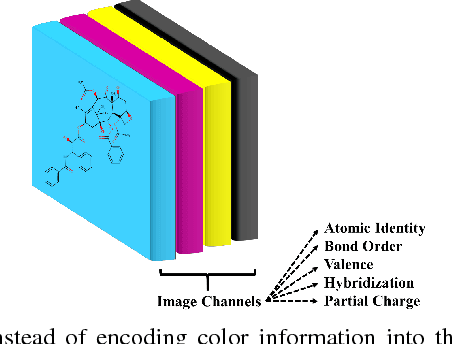
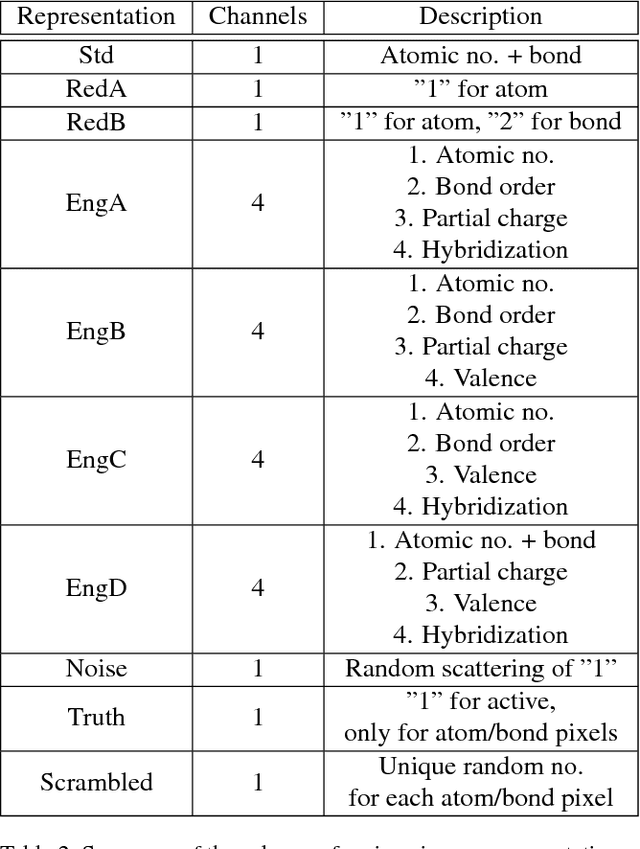
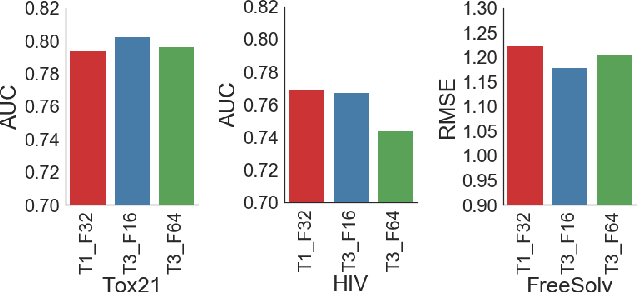
The meteoric rise of deep learning models in computer vision research, having achieved human-level accuracy in image recognition tasks is firm evidence of the impact of representation learning of deep neural networks. In the chemistry domain, recent advances have also led to the development of similar CNN models, such as Chemception, that is trained to predict chemical properties using images of molecular drawings. In this work, we investigate the effects of systematically removing and adding localized domain-specific information to the image channels of the training data. By augmenting images with only 3 additional basic information, and without introducing any architectural changes, we demonstrate that an augmented Chemception (AugChemception) outperforms the original model in the prediction of toxicity, activity, and solvation free energy. Then, by altering the information content in the images, and examining the resulting model's performance, we also identify two distinct learning patterns in predicting toxicity/activity as compared to solvation free energy. These patterns suggest that Chemception is learning about its tasks in the manner that is consistent with established knowledge. Thus, our work demonstrates that advanced chemical knowledge is not a pre-requisite for deep learning models to accurately predict complex chemical properties.
SMILES2Vec: An Interpretable General-Purpose Deep Neural Network for Predicting Chemical Properties
Mar 18, 2018
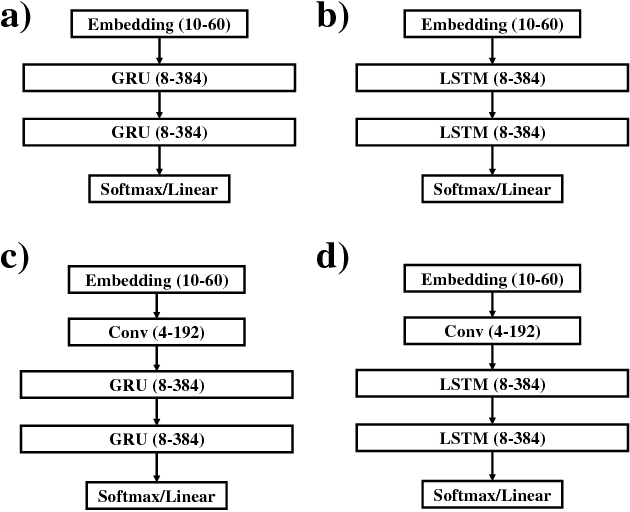
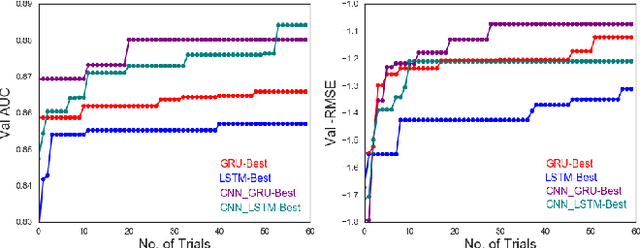
Chemical databases store information in text representations, and the SMILES format is a universal standard used in many cheminformatics software. Encoded in each SMILES string is structural information that can be used to predict complex chemical properties. In this work, we develop SMILES2vec, a deep RNN that automatically learns features from SMILES to predict chemical properties, without the need for additional explicit feature engineering. Using Bayesian optimization methods to tune the network architecture, we show that an optimized SMILES2vec model can serve as a general-purpose neural network for predicting distinct chemical properties including toxicity, activity, solubility and solvation energy, while also outperforming contemporary MLP neural networks that uses engineered features. Furthermore, we demonstrate proof-of-concept of interpretability by developing an explanation mask that localizes on the most important characters used in making a prediction. When tested on the solubility dataset, it identified specific parts of a chemical that is consistent with established first-principles knowledge with an accuracy of 88%. Our work demonstrates that neural networks can learn technically accurate chemical concept and provide state-of-the-art accuracy, making interpretable deep neural networks a useful tool of relevance to the chemical industry.
Using Rule-Based Labels for Weak Supervised Learning: A ChemNet for Transferable Chemical Property Prediction
Mar 18, 2018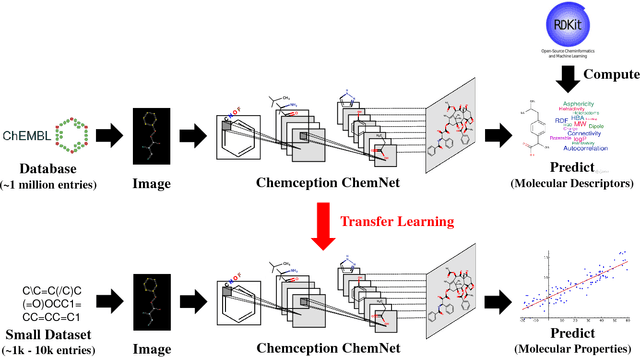
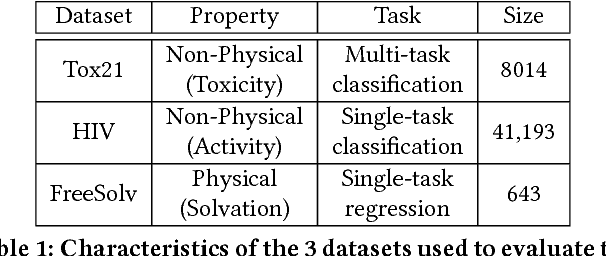
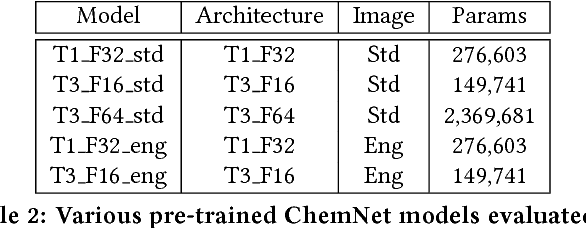
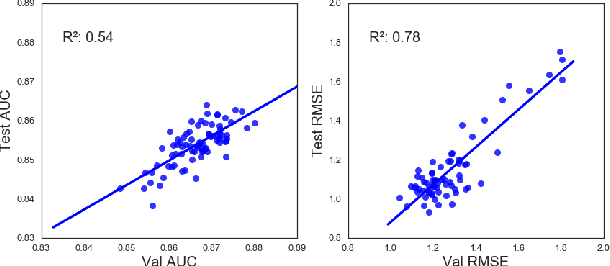
With access to large datasets, deep neural networks (DNN) have achieved human-level accuracy in image and speech recognition tasks. However, in chemistry, data is inherently small and fragmented. In this work, we develop an approach of using rule-based knowledge for training ChemNet, a transferable and generalizable deep neural network for chemical property prediction that learns in a weak-supervised manner from large unlabeled chemical databases. When coupled with transfer learning approaches to predict other smaller datasets for chemical properties that it was not originally trained on, we show that ChemNet's accuracy outperforms contemporary DNN models that were trained using conventional supervised learning. Furthermore, we demonstrate that the ChemNet pre-training approach is equally effective on both CNN (Chemception) and RNN (SMILES2vec) models, indicating that this approach is network architecture agnostic and is effective across multiple data modalities. Our results indicate a pre-trained ChemNet that incorporates chemistry domain knowledge, enables the development of generalizable neural networks for more accurate prediction of novel chemical properties.
GossipGraD: Scalable Deep Learning using Gossip Communication based Asynchronous Gradient Descent
Mar 15, 2018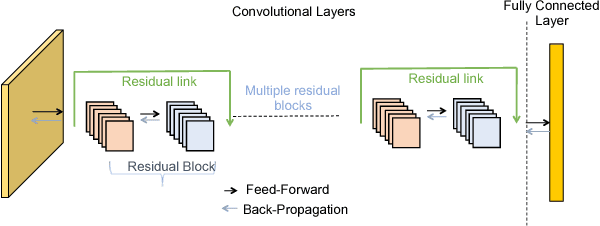
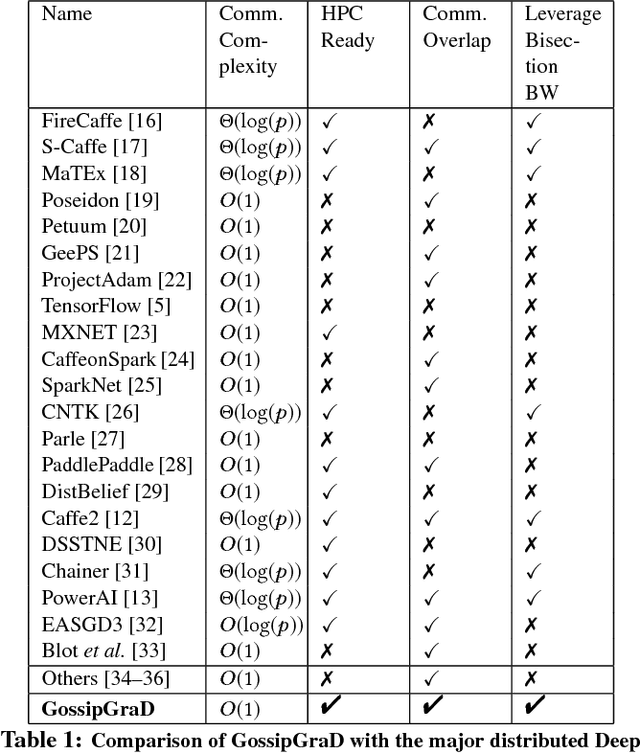
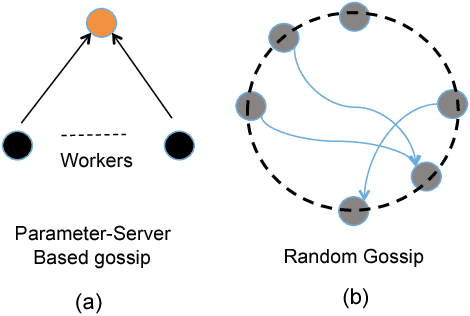

In this paper, we present GossipGraD - a gossip communication protocol based Stochastic Gradient Descent (SGD) algorithm for scaling Deep Learning (DL) algorithms on large-scale systems. The salient features of GossipGraD are: 1) reduction in overall communication complexity from {\Theta}(log(p)) for p compute nodes in well-studied SGD to O(1), 2) model diffusion such that compute nodes exchange their updates (gradients) indirectly after every log(p) steps, 3) rotation of communication partners for facilitating direct diffusion of gradients, 4) asynchronous distributed shuffle of samples during the feedforward phase in SGD to prevent over-fitting, 5) asynchronous communication of gradients for further reducing the communication cost of SGD and GossipGraD. We implement GossipGraD for GPU and CPU clusters and use NVIDIA GPUs (Pascal P100) connected with InfiniBand, and Intel Knights Landing (KNL) connected with Aries network. We evaluate GossipGraD using well-studied dataset ImageNet-1K (~250GB), and widely studied neural network topologies such as GoogLeNet and ResNet50 (current winner of ImageNet Large Scale Visualization Research Challenge (ILSVRC)). Our performance evaluation using both KNL and Pascal GPUs indicates that GossipGraD can achieve perfect efficiency for these datasets and their associated neural network topologies. Specifically, for ResNet50, GossipGraD is able to achieve ~100% compute efficiency using 128 NVIDIA Pascal P100 GPUs - while matching the top-1 classification accuracy published in literature.
Chemception: A Deep Neural Network with Minimal Chemistry Knowledge Matches the Performance of Expert-developed QSAR/QSPR Models
Jun 20, 2017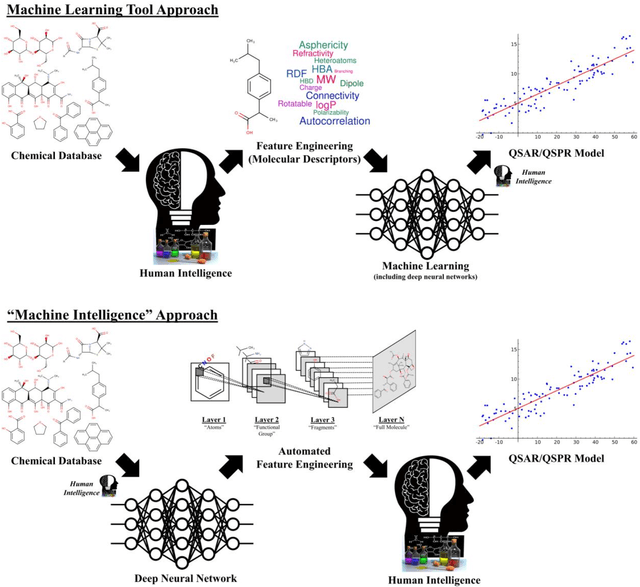
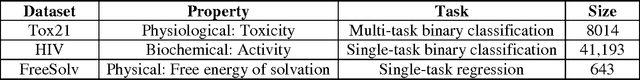
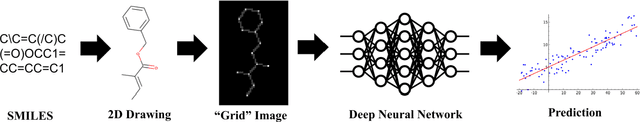
In the last few years, we have seen the transformative impact of deep learning in many applications, particularly in speech recognition and computer vision. Inspired by Google's Inception-ResNet deep convolutional neural network (CNN) for image classification, we have developed "Chemception", a deep CNN for the prediction of chemical properties, using just the images of 2D drawings of molecules. We develop Chemception without providing any additional explicit chemistry knowledge, such as basic concepts like periodicity, or advanced features like molecular descriptors and fingerprints. We then show how Chemception can serve as a general-purpose neural network architecture for predicting toxicity, activity, and solvation properties when trained on a modest database of 600 to 40,000 compounds. When compared to multi-layer perceptron (MLP) deep neural networks trained with ECFP fingerprints, Chemception slightly outperforms in activity and solvation prediction and slightly underperforms in toxicity prediction. Having matched the performance of expert-developed QSAR/QSPR deep learning models, our work demonstrates the plausibility of using deep neural networks to assist in computational chemistry research, where the feature engineering process is performed primarily by a deep learning algorithm.
Adaptive Neuron Apoptosis for Accelerating Deep Learning on Large Scale Systems
Oct 03, 2016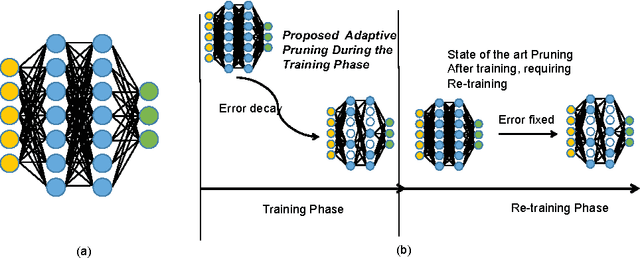
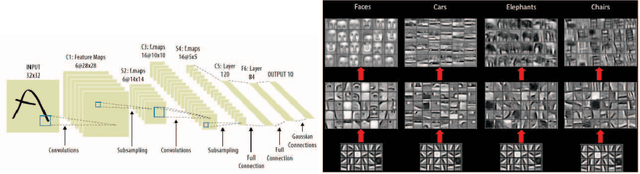
We present novel techniques to accelerate the convergence of Deep Learning algorithms by conducting low overhead removal of redundant neurons -- apoptosis of neurons -- which do not contribute to model learning, during the training phase itself. We provide in-depth theoretical underpinnings of our heuristics (bounding accuracy loss and handling apoptosis of several neuron types), and present the methods to conduct adaptive neuron apoptosis. Specifically, we are able to improve the training time for several datasets by 2-3x, while reducing the number of parameters by up to 30x (4-5x on average) on datasets such as ImageNet classification. For the Higgs Boson dataset, our implementation improves the accuracy (measured by Area Under Curve (AUC)) for classification from 0.88/1 to 0.94/1, while reducing the number of parameters by 3x in comparison to existing literature. The proposed methods achieve a 2.44x speedup in comparison to the default (no apoptosis) algorithm.