 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMLPerf HPC: A Holistic Benchmark Suite for Scientific Machine Learning on HPC Systems
Oct 26, 2021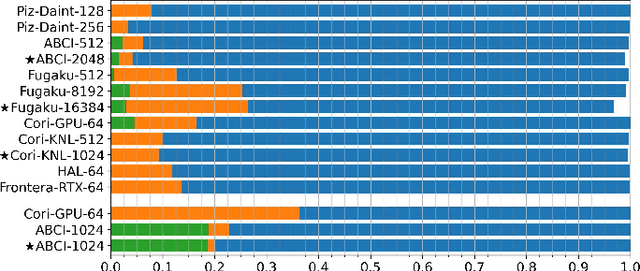
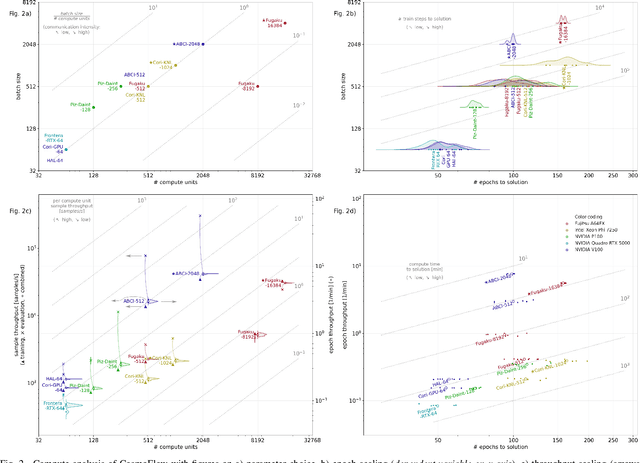
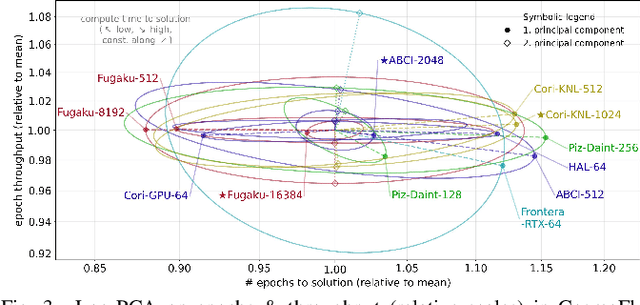
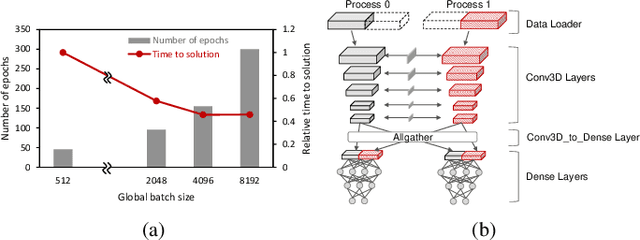
Scientific communities are increasingly adopting machine learning and deep learning models in their applications to accelerate scientific insights. High performance computing systems are pushing the frontiers of performance with a rich diversity of hardware resources and massive scale-out capabilities. There is a critical need to understand fair and effective benchmarking of machine learning applications that are representative of real-world scientific use cases. MLPerf is a community-driven standard to benchmark machine learning workloads, focusing on end-to-end performance metrics. In this paper, we introduce MLPerf HPC, a benchmark suite of large-scale scientific machine learning training applications driven by the MLCommons Association. We present the results from the first submission round, including a diverse set of some of the world's largest HPC systems. We develop a systematic framework for their joint analysis and compare them in terms of data staging, algorithmic convergence, and compute performance. As a result, we gain a quantitative understanding of optimizations on different subsystems such as staging and on-node loading of data, compute-unit utilization, and communication scheduling, enabling overall $>10 \times$ (end-to-end) performance improvements through system scaling. Notably, our analysis shows a scale-dependent interplay between the dataset size, a system's memory hierarchy, and training convergence that underlines the importance of near-compute storage. To overcome the data-parallel scalability challenge at large batch sizes, we discuss specific learning techniques and hybrid data-and-model parallelism that are effective on large systems. We conclude by characterizing each benchmark with respect to low-level memory, I/O, and network behavior to parameterize extended roofline performance models in future rounds.
PharML.Bind: Pharmacologic Machine Learning for Protein-Ligand Interactions
Oct 23, 2019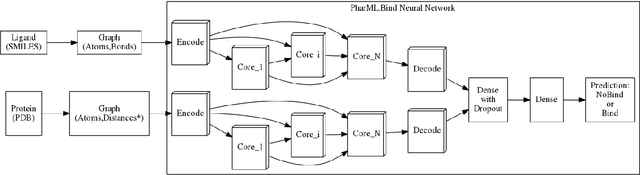
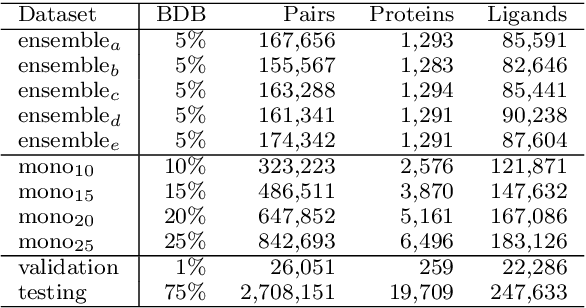

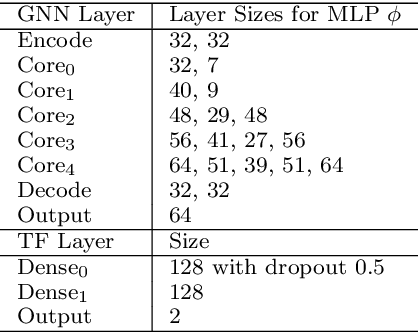
Is it feasible to create an analysis paradigm that can analyze and then accurately and quickly predict known drugs from experimental data? PharML.Bind is a machine learning toolkit which is able to accomplish this feat. Utilizing deep neural networks and big data, PharML.Bind correlates experimentally-derived drug affinities and protein-ligand X-ray structures to create novel predictions. The utility of PharML.Bind is in its application as a rapid, accurate, and robust prediction platform for discovery and personalized medicine. This paper demonstrates that graph neural networks (GNNs) can be trained to screen hundreds of thousands of compounds against thousands of targets in minutes, a vastly shorter time than previous approaches. This manuscript presents results from training and testing using the entirety of BindingDB after cleaning; this includes a test set with 19,708 X-ray structures and 247,633 drugs, leading to 2,708,151 unique protein-ligand pairings. PharML.Bind achieves a prodigious 98.3% accuracy on this test set in under 25 minutes. PharML.Bind is premised on the following key principles: 1) speed and a high enrichment factor per unit compute time, provided by high-quality training data combined with a novel GNN architecture and use of high-performance computing resources, 2) the ability to generalize to proteins and drugs outside of the training set, including those with unknown active sites, through the use of an active-site-agnostic GNN mapping, and 3) the ability to be easily integrated as a component of increasingly-complex prediction and analysis pipelines. PharML.Bind represents a timely and practical approach to leverage the power of machine learning to efficiently analyze and predict drug action on any practical scale and will provide utility in a variety of discovery and medical applications.
Recombination of Artificial Neural Networks
Jan 12, 2019
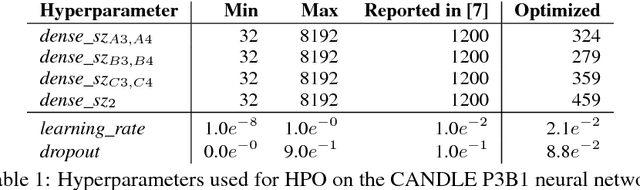
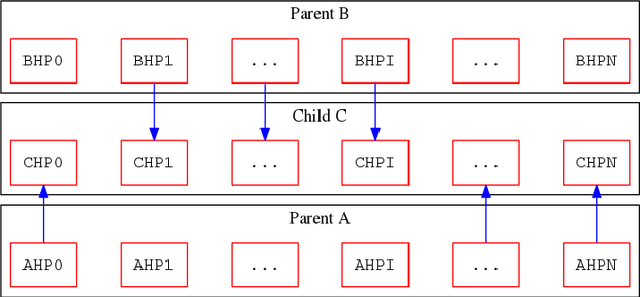

We propose a genetic algorithm (GA) for hyperparameter optimization of artificial neural networks which includes chromosomal crossover as well as a decoupling of parameters (i.e., weights and biases) from hyperparameters (e.g., learning rate, weight decay, and dropout) during sexual reproduction. Children are produced from three parents; two contributing hyperparameters and one contributing the parameters. Our version of population-based training (PBT) combines traditional gradient-based approaches such as stochastic gradient descent (SGD) with our GA to optimize both parameters and hyperparameters across SGD epochs. Our improvements over traditional PBT provide an increased speed of adaptation and a greater ability to shed deleterious genes from the population. Our methods improve final accuracy as well as time to fixed accuracy on a wide range of deep neural network architectures including convolutional neural networks, recurrent neural networks, dense neural networks, and capsule networks.