 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePharML.Bind: Pharmacologic Machine Learning for Protein-Ligand Interactions
Paper and Code
Oct 23, 2019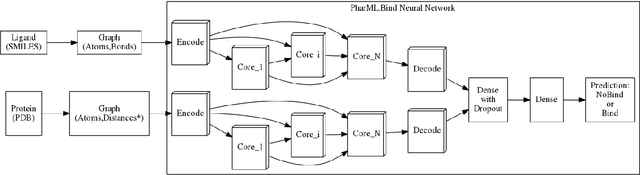
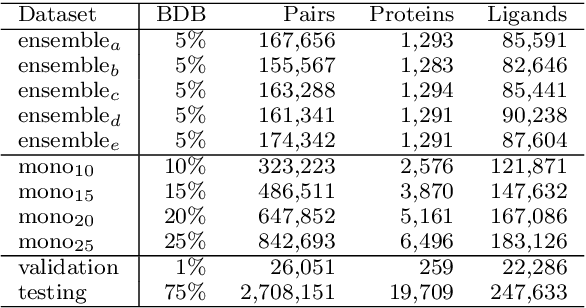

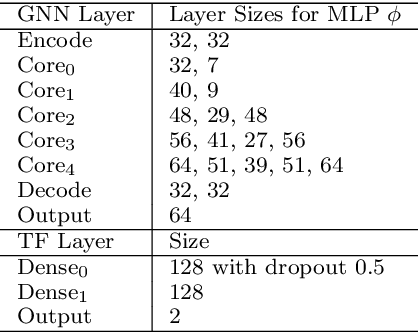
Is it feasible to create an analysis paradigm that can analyze and then accurately and quickly predict known drugs from experimental data? PharML.Bind is a machine learning toolkit which is able to accomplish this feat. Utilizing deep neural networks and big data, PharML.Bind correlates experimentally-derived drug affinities and protein-ligand X-ray structures to create novel predictions. The utility of PharML.Bind is in its application as a rapid, accurate, and robust prediction platform for discovery and personalized medicine. This paper demonstrates that graph neural networks (GNNs) can be trained to screen hundreds of thousands of compounds against thousands of targets in minutes, a vastly shorter time than previous approaches. This manuscript presents results from training and testing using the entirety of BindingDB after cleaning; this includes a test set with 19,708 X-ray structures and 247,633 drugs, leading to 2,708,151 unique protein-ligand pairings. PharML.Bind achieves a prodigious 98.3% accuracy on this test set in under 25 minutes. PharML.Bind is premised on the following key principles: 1) speed and a high enrichment factor per unit compute time, provided by high-quality training data combined with a novel GNN architecture and use of high-performance computing resources, 2) the ability to generalize to proteins and drugs outside of the training set, including those with unknown active sites, through the use of an active-site-agnostic GNN mapping, and 3) the ability to be easily integrated as a component of increasingly-complex prediction and analysis pipelines. PharML.Bind represents a timely and practical approach to leverage the power of machine learning to efficiently analyze and predict drug action on any practical scale and will provide utility in a variety of discovery and medical applications.