 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePhylogeny-Informed Interaction Estimation Accelerates Co-Evolutionary Learning
Apr 09, 2024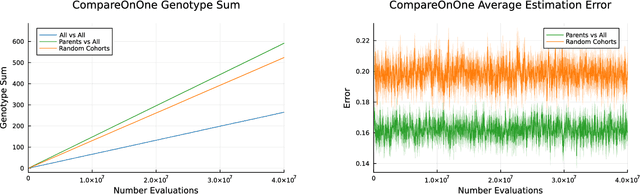
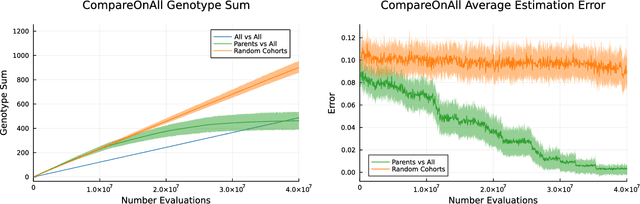
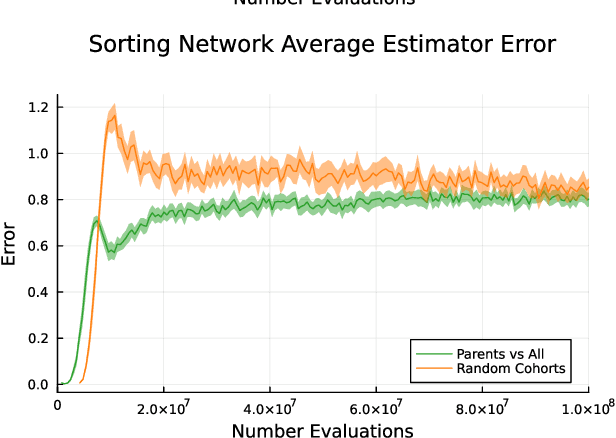
Co-evolution is a powerful problem-solving approach. However, fitness evaluation in co-evolutionary algorithms can be computationally expensive, as the quality of an individual in one population is defined by its interactions with many (or all) members of one or more other populations. To accelerate co-evolutionary systems, we introduce phylogeny-informed interaction estimation, which uses runtime phylogenetic analysis to estimate interaction outcomes between individuals based on how their relatives performed against each other. We test our interaction estimation method with three distinct co-evolutionary systems: two systems focused on measuring problem-solving success and one focused on measuring evolutionary open-endedness. We find that phylogeny-informed estimation can substantially reduce the computation required to solve problems, particularly at the beginning of long-term evolutionary runs. Additionally, we find that our estimation method initially jump-starts the evolution of neural complexity in our open-ended domain, but estimation-free systems eventually "catch-up" if given enough time. More broadly, continued refinements to these phylogeny-informed interaction estimation methods offers a promising path to reducing the computational cost of running co-evolutionary systems while maintaining their open-endedness.
Runtime phylogenetic analysis enables extreme subsampling for test-based problems
Feb 02, 2024



A phylogeny describes the evolutionary history of an evolving population. Evolutionary search algorithms can perfectly track the ancestry of candidate solutions, illuminating a population's trajectory through the search space. However, phylogenetic analyses are typically limited to post-hoc studies of search performance. We introduce phylogeny-informed subsampling, a new class of subsampling methods that exploit runtime phylogenetic analyses for solving test-based problems. Specifically, we assess two phylogeny-informed subsampling methods -- individualized random subsampling and ancestor-based subsampling -- on three diagnostic problems and ten genetic programming (GP) problems from program synthesis benchmark suites. Overall, we found that phylogeny-informed subsampling methods enable problem-solving success at extreme subsampling levels where other subsampling methods fail. For example, phylogeny-informed subsampling methods more reliably solved program synthesis problems when evaluating just one training case per-individual, per-generation. However, at moderate subsampling levels, phylogeny-informed subsampling generally performed no better than random subsampling on GP problems. Our diagnostic experiments show that phylogeny-informed subsampling improves diversity maintenance relative to random subsampling, but its effects on a selection scheme's capacity to rapidly exploit fitness gradients varied by selection scheme. Continued refinements of phylogeny-informed subsampling techniques offer a promising new direction for scaling up evolutionary systems to handle problems with many expensive-to-evaluate fitness criteria.
Reachability Analysis for Lexicase Selection via Community Assembly Graphs
Sep 20, 2023Fitness landscapes have historically been a powerful tool for analyzing the search space explored by evolutionary algorithms. In particular, they facilitate understanding how easily reachable an optimal solution is from a given starting point. However, simple fitness landscapes are inappropriate for analyzing the search space seen by selection schemes like lexicase selection in which the outcome of selection depends heavily on the current contents of the population (i.e. selection schemes with complex ecological dynamics). Here, we propose borrowing a tool from ecology to solve this problem: community assembly graphs. We demonstrate a simple proof-of-concept for this approach on an NK Landscape where we have perfect information. We then demonstrate that this approach can be successfully applied to a complex genetic programming problem. While further research is necessary to understand how to best use this tool, we believe it will be a valuable addition to our toolkit and facilitate analyses that were previously impossible.
Phylogeny-informed fitness estimation
Jun 06, 2023



Phylogenies (ancestry trees) depict the evolutionary history of an evolving population. In evolutionary computing, a phylogeny can reveal how an evolutionary algorithm steers a population through a search space, illuminating the step-by-step process by which any solutions evolve. Thus far, phylogenetic analyses have primarily been applied as post-hoc analyses used to deepen our understanding of existing evolutionary algorithms. Here, we investigate whether phylogenetic analyses can be used at runtime to augment parent selection procedures during an evolutionary search. Specifically, we propose phylogeny-informed fitness estimation, which exploits a population's phylogeny to estimate fitness evaluations. We evaluate phylogeny-informed fitness estimation in the context of the down-sampled lexicase and cohort lexicase selection algorithms on two diagnostic analyses and four genetic programming (GP) problems. Our results indicate that phylogeny-informed fitness estimation can mitigate the drawbacks of down-sampled lexicase, improving diversity maintenance and search space exploration. However, the extent to which phylogeny-informed fitness estimation improves problem-solving success for GP varies by problem, subsampling method, and subsampling level. This work serves as an initial step toward improving evolutionary algorithms by exploiting runtime phylogenetic analysis.
A Static Analysis of Informed Down-Samples
Apr 17, 2023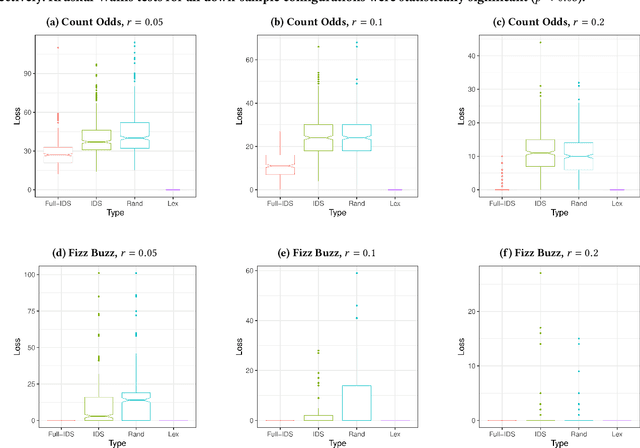
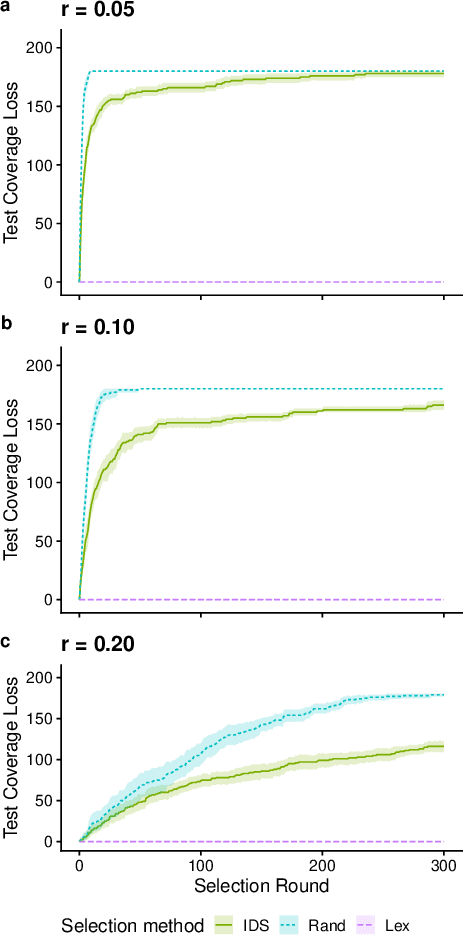
We present an analysis of the loss of population-level test coverage induced by different down-sampling strategies when combined with lexicase selection. We study recorded populations from the first generation of genetic programming runs, as well as entirely synthetic populations. Our findings verify the hypothesis that informed down-sampling better maintains population-level test coverage when compared to random down-sampling. Additionally, we show that both forms of down-sampling cause greater test coverage loss than standard lexicase selection with no down-sampling. However, given more information about the population, we found that informed down-sampling can further reduce its test coverage loss. We also recommend wider adoption of the static population analyses we present in this work.
Analyzing the Interaction Between Down-Sampling and Selection
Apr 14, 2023Genetic programming systems often use large training sets to evaluate the quality of candidate solutions for selection. However, evaluating populations on large training sets can be computationally expensive. Down-sampling training sets has long been used to decrease the computational cost of evaluation in a wide range of application domains. Indeed, recent studies have shown that both random and informed down-sampling can substantially improve problem-solving success for GP systems that use the lexicase parent selection algorithm. We use the PushGP framework to experimentally test whether these down-sampling techniques can also improve problem-solving success in the context of two other commonly used selection methods, fitness-proportionate and tournament selection, across eight GP problems (four program synthesis and four symbolic regression). We verified that down-sampling can benefit the problem-solving success of both fitness-proportionate and tournament selection. However, the number of problems wherein down-sampling improved problem-solving success varied by selection scheme, suggesting that the impact of down-sampling depends both on the problem and choice of selection scheme. Surprisingly, we found that down-sampling was most consistently beneficial when combined with lexicase selection as compared to tournament and fitness-proportionate selection. Overall, our results suggest that down-sampling should be considered more often when solving test-based GP problems.
Informed Down-Sampled Lexicase Selection: Identifying productive training cases for efficient problem solving
Jan 04, 2023



Genetic Programming (GP) often uses large training sets and requires all individuals to be evaluated on all training cases during selection. Random down-sampled lexicase selection evaluates individuals on only a random subset of the training cases allowing for more individuals to be explored with the same amount of program executions. However, creating a down-sample randomly might exclude important cases from the current down-sample for a number of generations, while cases that measure the same behavior (synonymous cases) may be overused despite their redundancy. In this work, we introduce Informed Down-Sampled Lexicase Selection. This method leverages population statistics to build down-samples that contain more distinct and therefore informative training cases. Through an empirical investigation across two different GP systems (PushGP and Grammar-Guided GP), we find that informed down-sampling significantly outperforms random down-sampling on a set of contemporary program synthesis benchmark problems. Through an analysis of the created down-samples, we find that important training cases are included in the down-sample consistently across independent evolutionary runs and systems. We hypothesize that this improvement can be attributed to the ability of Informed Down-Sampled Lexicase Selection to maintain more specialist individuals over the course of evolution, while also benefiting from reduced per-evaluation costs.
A suite of diagnostic metrics for characterizing selection schemes
Apr 29, 2022


Evolutionary algorithms are effective general-purpose techniques for solving optimization problems. Understanding how each component of an evolutionary algorithm influences its problem-solving success improves our ability to target particular problem domains. Our work focuses on evaluating selection schemes, which choose individuals to contribute genetic material to the next generation. We introduce four diagnostic search spaces for testing the strengths and weaknesses of selection schemes: the exploitation rate diagnostic, ordered exploitation rate diagnostic, contradictory objectives diagnostic, and the multi-path exploration diagnostic. Each diagnostic is handcrafted to isolate and measure the relative exploitation and exploration characteristics of selection schemes. In this study, we use our diagnostics to evaluate six population selection methods: truncation selection, tournament selection, fitness sharing, lexicase selection, nondominated sorting, and novelty search. Expectedly, tournament and truncation selection excelled in gradient exploitation but poorly explored search spaces, and novelty search excelled at exploration but failed to exploit fitness gradients. Fitness sharing performed poorly across all diagnostics, suggesting poor overall exploitation and exploration abilities. Nondominated sorting was best for maintaining populations comprised of individuals with different trade-offs of multiple objectives, but struggled to effectively exploit fitness gradients. Lexicase selection balanced search space exploration with exploitation, generally performing well across diagnostics. Our work demonstrates the value of diagnostic search spaces for building a deeper understanding of selection schemes, which can then be used to improve or develop new selection methods.
What can phylogenetic metrics tell us about useful diversity in evolutionary algorithms?
Aug 28, 2021
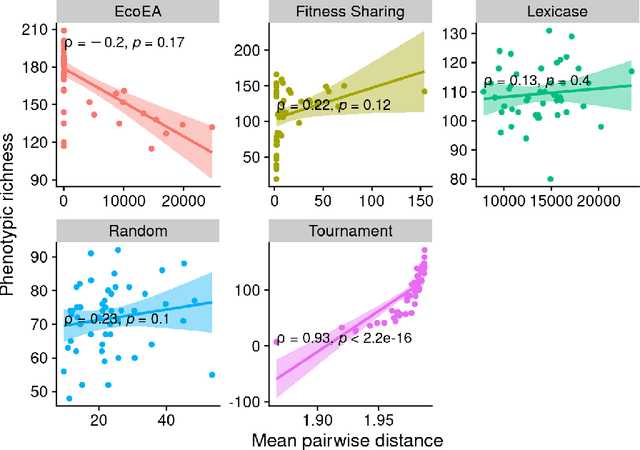
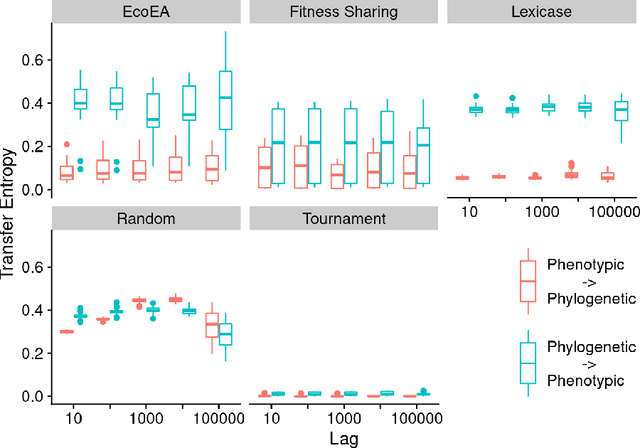
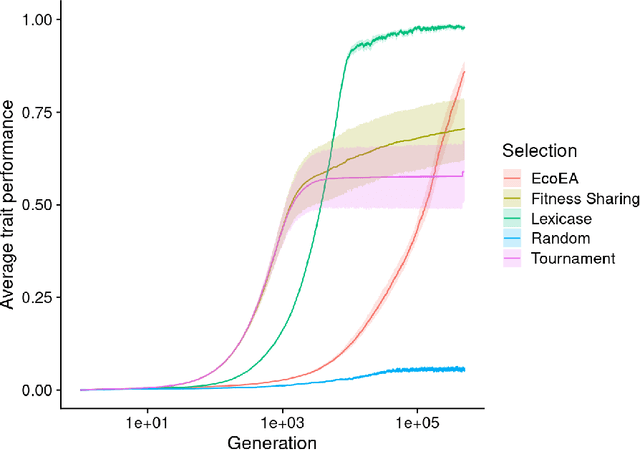
It is generally accepted that "diversity" is associated with success in evolutionary algorithms. However, diversity is a broad concept that can be measured and defined in a multitude of ways. To date, most evolutionary computation research has measured diversity using the richness and/or evenness of a particular genotypic or phenotypic property. While these metrics are informative, we hypothesize that other diversity metrics are more strongly predictive of success. Phylogenetic diversity metrics are a class of metrics popularly used in biology, which take into account the evolutionary history of a population. Here, we investigate the extent to which 1) these metrics provide different information than those traditionally used in evolutionary computation, and 2) these metrics better predict the long-term success of a run of evolutionary computation. We find that, in most cases, phylogenetic metrics behave meaningfully differently from other diversity metrics. Moreover, our results suggest that phylogenetic diversity is indeed a better predictor of success.
Matchmaker, Matchmaker, Make Me a Match: Geometric, Variational, and Evolutionary Implications of Criteria for Tag Affinity
Aug 10, 2021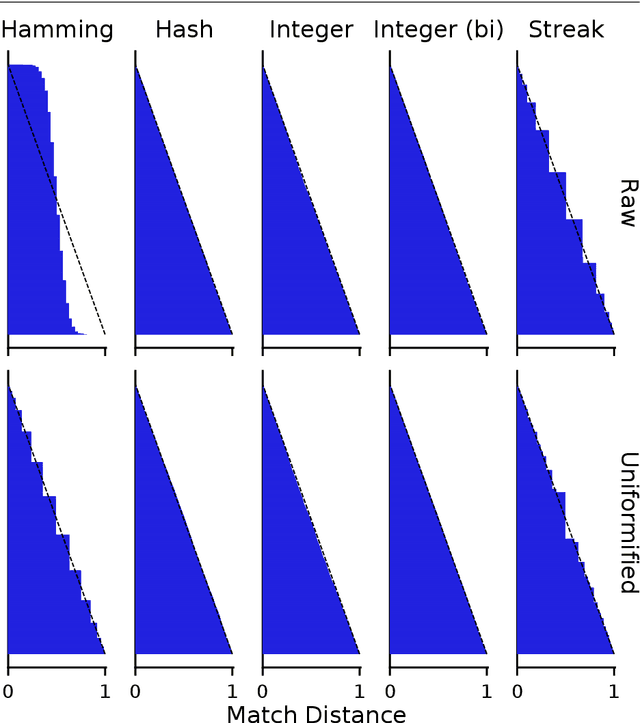

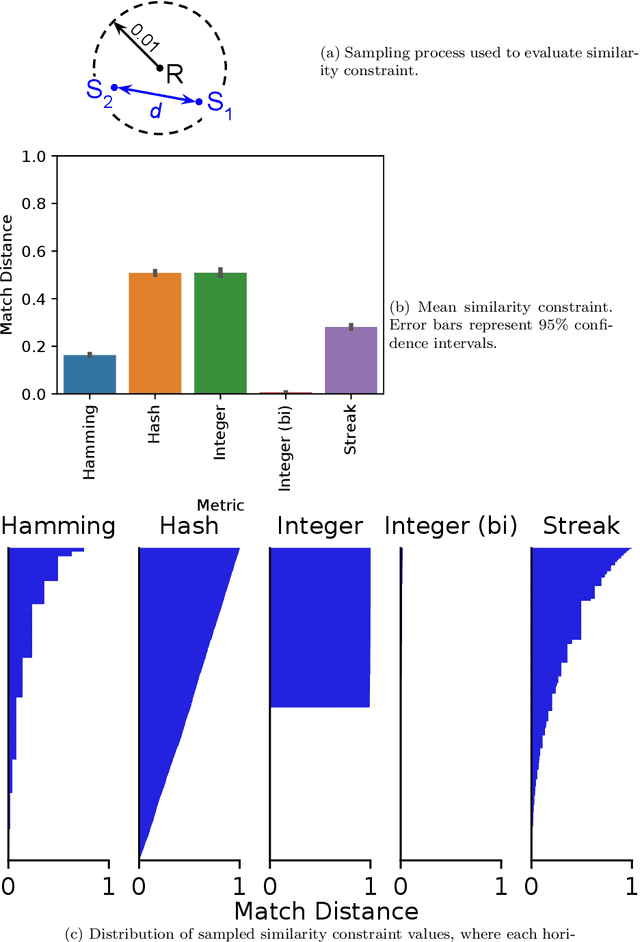
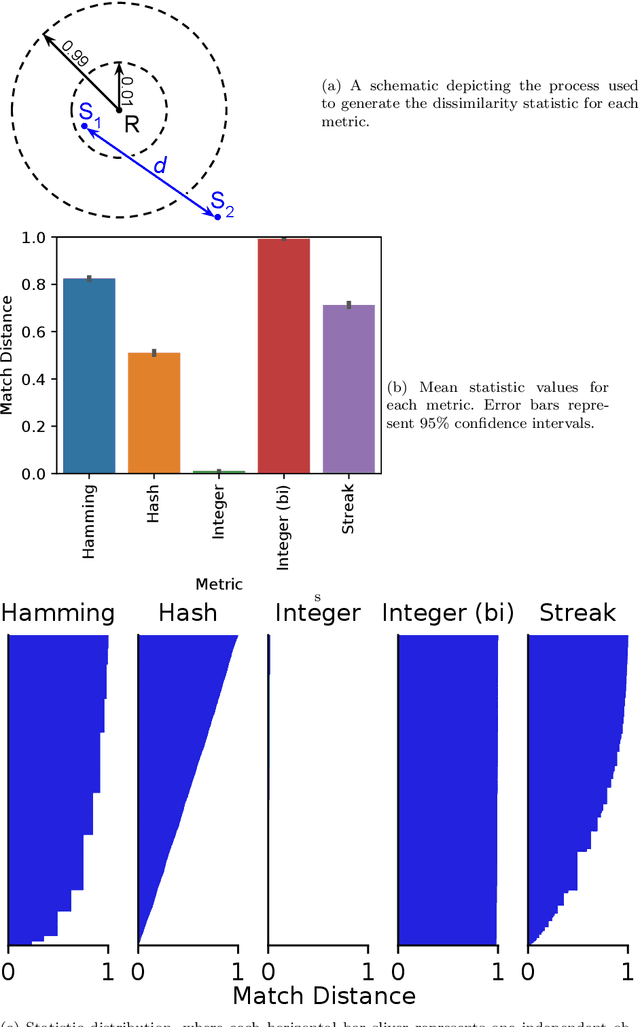
Genetic programming and artificial life systems commonly employ tag-matching schemes to determine interactions between model components. However, the implications of criteria used to determine affinity between tags with respect to constraints on emergent connectivity, canalization of changes to connectivity under mutation, and evolutionary dynamics have not been considered. We highlight differences between tag-matching criteria with respect to geometric constraint and variation generated under mutation. We find that tag-matching criteria can influence the rate of adaptive evolution and the quality of evolved solutions. Better understanding of the geometric, variational, and evolutionary properties of tag-matching criteria will facilitate more effective incorporation of tag matching into genetic programming and artificial life systems. By showing that tag-matching criteria influence connectivity patterns and evolutionary dynamics, our findings also raise fundamental questions about the properties of tag-matching systems in nature.