 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024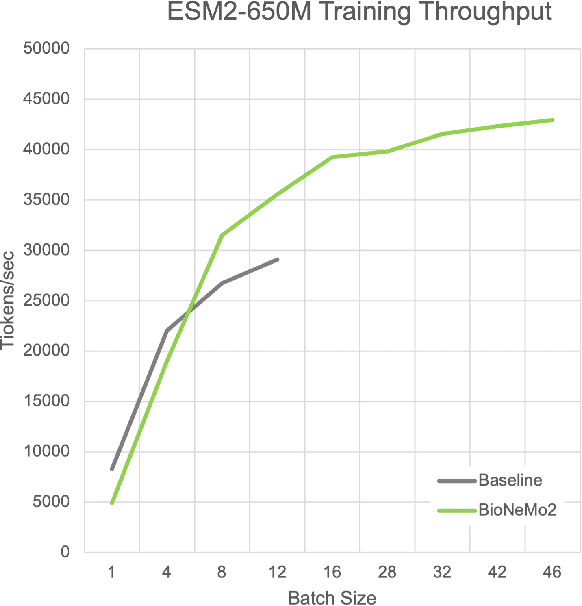
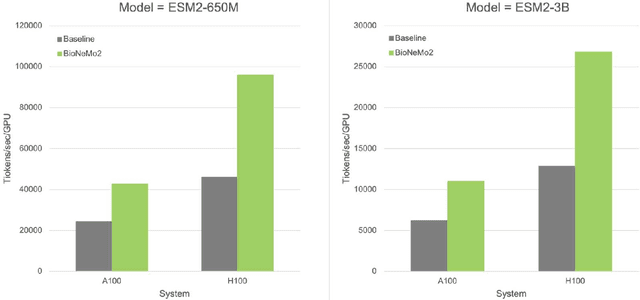
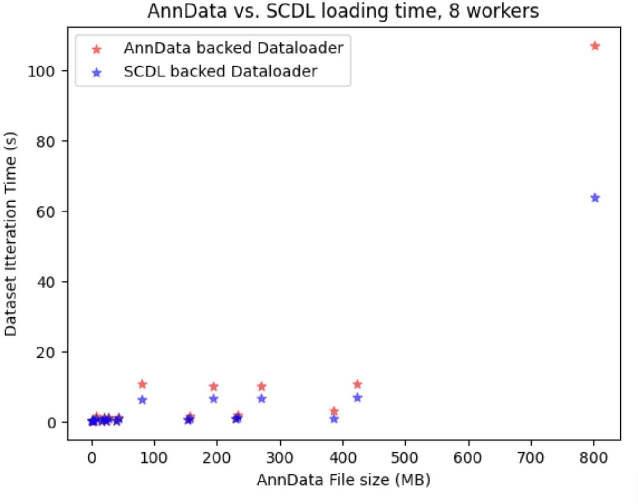
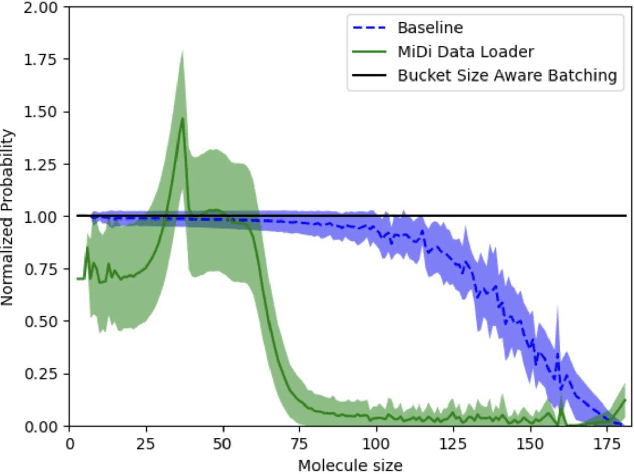
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
A view of Estimation of Distribution Algorithms through the lens of Expectation-Maximization
Jun 05, 2019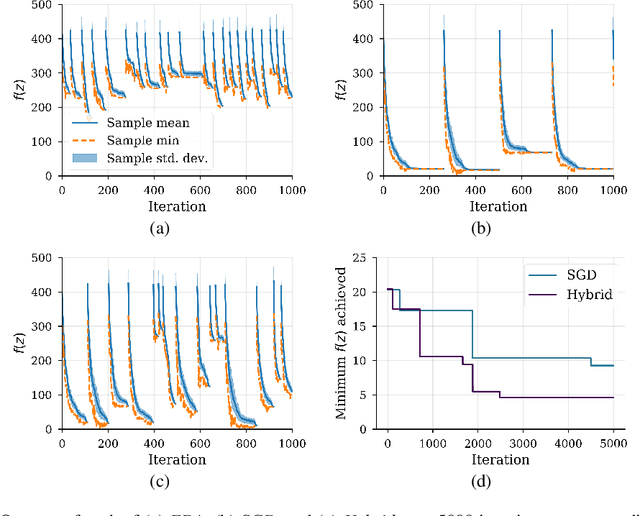
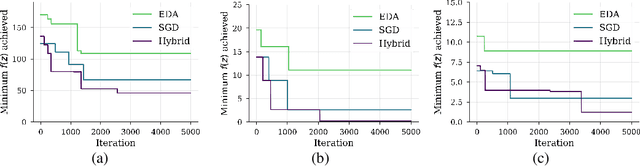
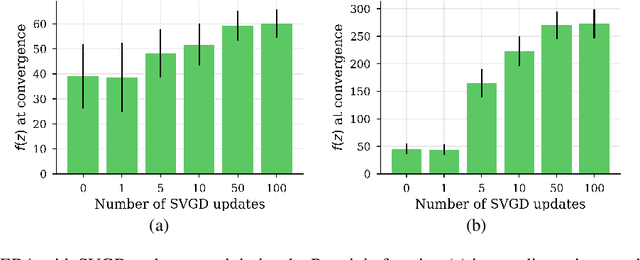
We show that under mild conditions, Estimation of Distribution Algorithms (EDAs) can be written as variational Expectation-Maximization (EM) that uses a mixture of weighted particles as the approximate posterior. In the infinite particle limit, EDAs can be viewed as exact EM. Because EM sits on a rigorous statistical foundation and has been thoroughly analyzed, this connection provides a coherent framework with which to reason about EDAs. Importantly, the connection also suggests avenues for possible improvements to EDAs owing to our ability to leverage general statistical tools and generalizations of EM. For example, we make use of results about known EM convergence properties to propose an adaptive, hybrid EDA-gradient descent algorithm; this hybrid demonstrates better performance than either component of the hybrid on several canonical, non-convex test functions. We also demonstrate empirically that although one might hypothesize that reducing the variational gap could prove useful, it actually degrades performance of EDAs. Finally, we show that the connection between EM and EDAs provides us with a new perspective on why EDAs are performing approximate natural gradient descent.
Next-Step Conditioned Deep Convolutional Neural Networks Improve Protein Secondary Structure Prediction
Feb 13, 2017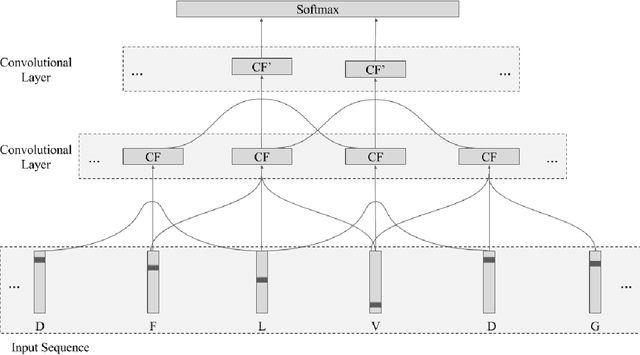
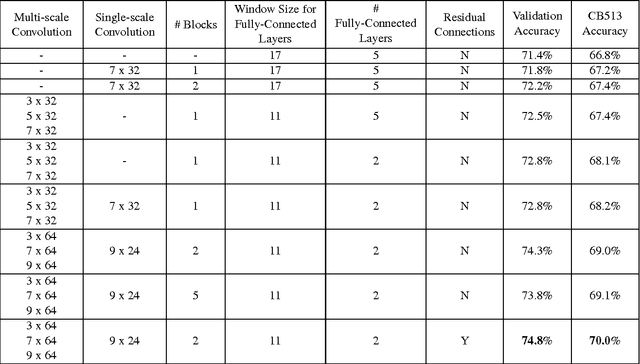
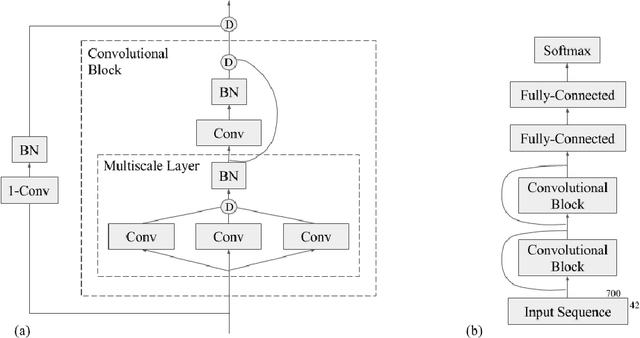
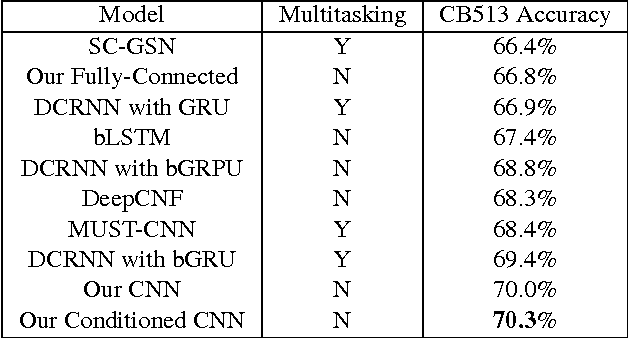
Recently developed deep learning techniques have significantly improved the accuracy of various speech and image recognition systems. In this paper we show how to adapt some of these techniques to create a novel chained convolutional architecture with next-step conditioning for improving performance on protein sequence prediction problems. We explore its value by demonstrating its ability to improve performance on eight-class secondary structure prediction. We first establish a state-of-the-art baseline by adapting recent advances in convolutional neural networks which were developed for vision tasks. This model achieves 70.0% per amino acid accuracy on the CB513 benchmark dataset without use of standard performance-boosting techniques such as ensembling or multitask learning. We then improve upon this state-of-the-art result using a novel chained prediction approach which frames the secondary structure prediction as a next-step prediction problem. This sequential model achieves 70.3% Q8 accuracy on CB513 with a single model; an ensemble of these models produces 71.4% Q8 accuracy on the same test set, improving upon the previous overall state of the art for the eight-class secondary structure problem. Our models are implemented using TensorFlow, an open-source machine learning software library available at TensorFlow.org; we aim to release the code for these experiments as part of the TensorFlow repository.
Protein Secondary Structure Prediction Using Deep Multi-scale Convolutional Neural Networks and Next-Step Conditioning
Nov 04, 2016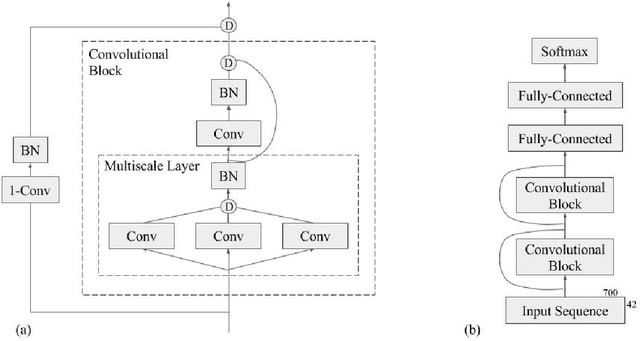

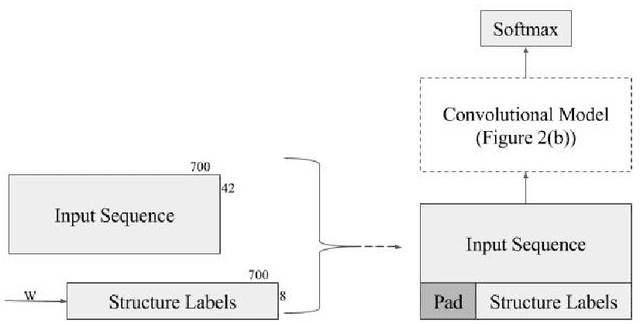
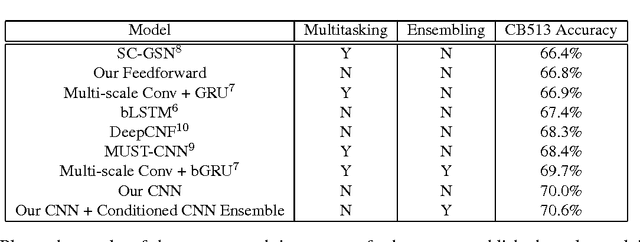
Recently developed deep learning techniques have significantly improved the accuracy of various speech and image recognition systems. In this paper we adapt some of these techniques for protein secondary structure prediction. We first train a series of deep neural networks to predict eight-class secondary structure labels given a protein's amino acid sequence information and find that using recent methods for regularization, such as dropout and weight-norm constraining, leads to measurable gains in accuracy. We then adapt recent convolutional neural network architectures--Inception, ReSNet, and DenseNet with Batch Normalization--to the problem of protein structure prediction. These convolutional architectures make heavy use of multi-scale filter layers that simultaneously compute features on several scales, and use residual connections to prevent underfitting. Using a carefully modified version of these architectures, we achieve state-of-the-art performance of 70.0% per amino acid accuracy on the public CB513 benchmark dataset. Finally, we explore additions from sequence-to-sequence learning, altering the model to make its predictions conditioned on both the protein's amino acid sequence and its past secondary structure labels. We introduce a new method of ensembling such a conditional model with our convolutional model, an approach which reaches 70.6% Q8 accuracy on CB513. We argue that these results can be further refined for larger boosts in prediction accuracy through more sophisticated attempts to control overfitting of conditional models. We aim to release the code for these experiments as part of the TensorFlow repository.