 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Lightweight U-like Network Utilizing Neural Memory Ordinary Differential Equations for Slimming the Decoder
Dec 09, 2024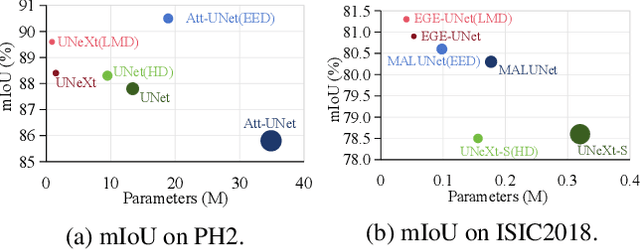
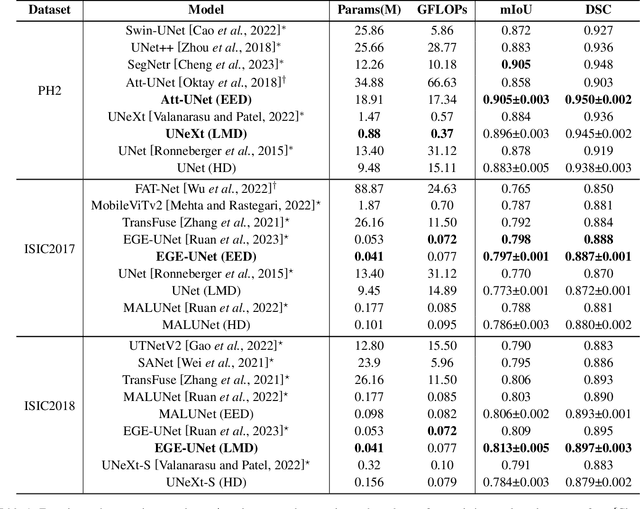
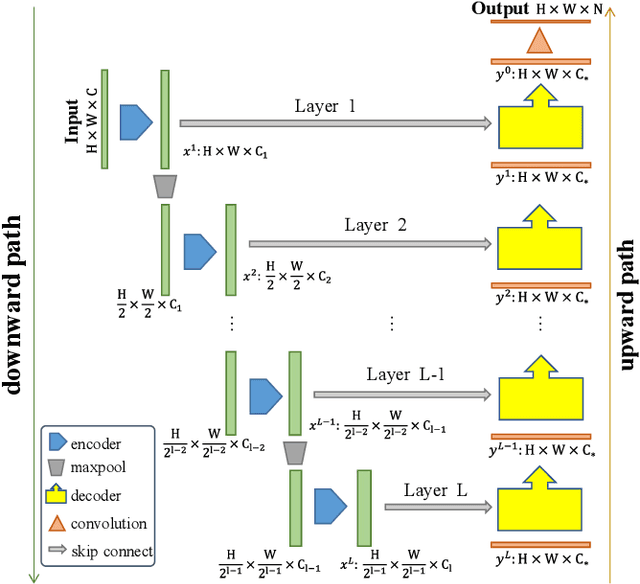
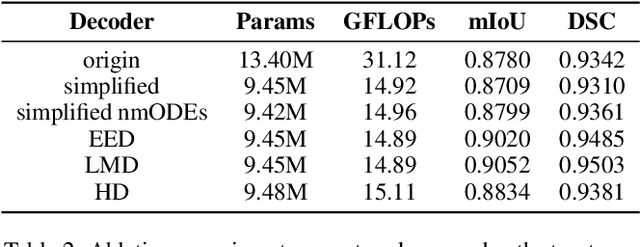
In recent years, advanced U-like networks have demonstrated remarkable performance in medical image segmentation tasks. However, their drawbacks, including excessive parameters, high computational complexity, and slow inference speed, pose challenges for practical implementation in scenarios with limited computational resources. Existing lightweight U-like networks have alleviated some of these problems, but they often have pre-designed structures and consist of inseparable modules, limiting their application scenarios. In this paper, we propose three plug-and-play decoders by employing different discretization methods of the neural memory Ordinary Differential Equations (nmODEs). These decoders integrate features at various levels of abstraction by processing information from skip connections and performing numerical operations on upward path. Through experiments on the PH2, ISIC2017, and ISIC2018 datasets, we embed these decoders into different U-like networks, demonstrating their effectiveness in significantly reducing the number of parameters and FLOPs while maintaining performance. In summary, the proposed discretized nmODEs decoders are capable of reducing the number of parameters by about 20% ~ 50% and FLOPs by up to 74%, while possessing the potential to adapt to all U-like networks. Our code is available at https://github.com/nayutayuki/Lightweight-nmODE-Decoders-For-U-like-networks.
Learn molecular representations from large-scale unlabeled molecules for drug discovery
Dec 21, 2020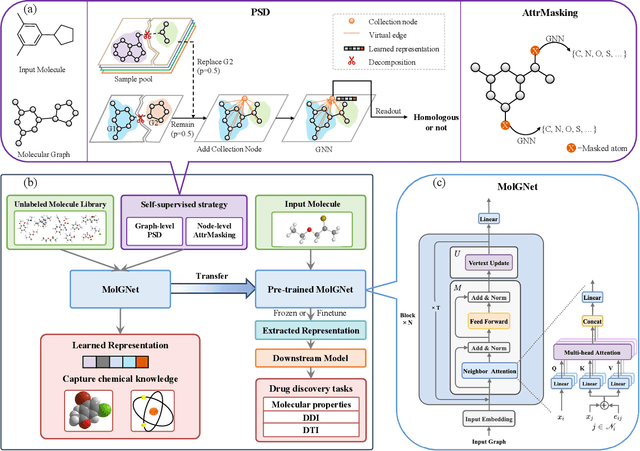

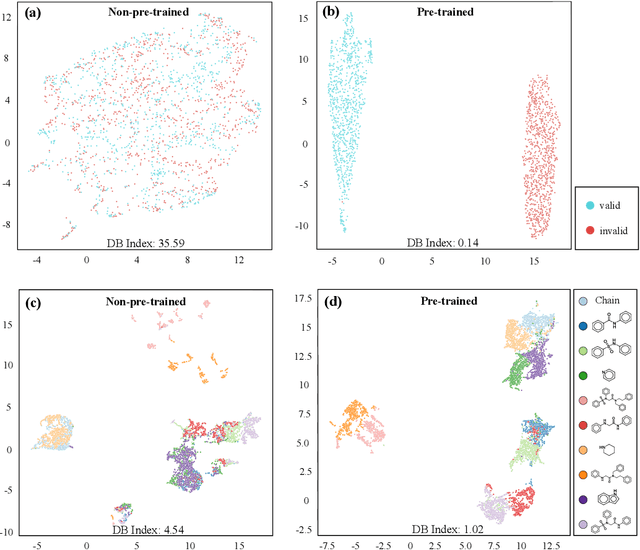

How to produce expressive molecular representations is a fundamental challenge in AI-driven drug discovery. Graph neural network (GNN) has emerged as a powerful technique for modeling molecular data. However, previous supervised approaches usually suffer from the scarcity of labeled data and have poor generalization capability. Here, we proposed a novel Molecular Pre-training Graph-based deep learning framework, named MPG, that leans molecular representations from large-scale unlabeled molecules. In MPG, we proposed a powerful MolGNet model and an effective self-supervised strategy for pre-training the model at both the node and graph-level. After pre-training on 11 million unlabeled molecules, we revealed that MolGNet can capture valuable chemistry insights to produce interpretable representation. The pre-trained MolGNet can be fine-tuned with just one additional output layer to create state-of-the-art models for a wide range of drug discovery tasks, including molecular properties prediction, drug-drug interaction, and drug-target interaction, involving 13 benchmark datasets. Our work demonstrates that MPG is promising to become a novel approach in the drug discovery pipeline.