 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearn molecular representations from large-scale unlabeled molecules for drug discovery
Dec 21, 2020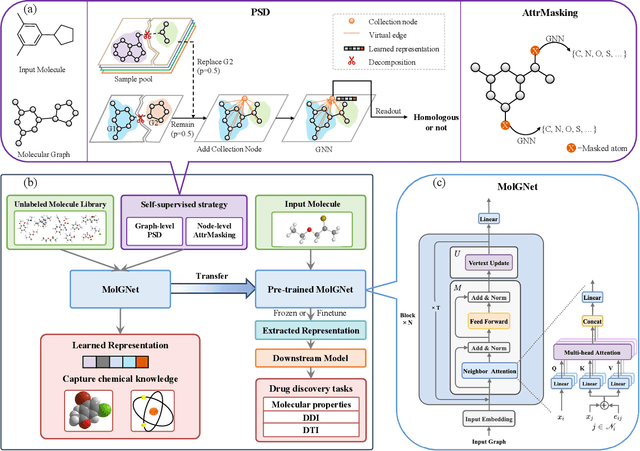

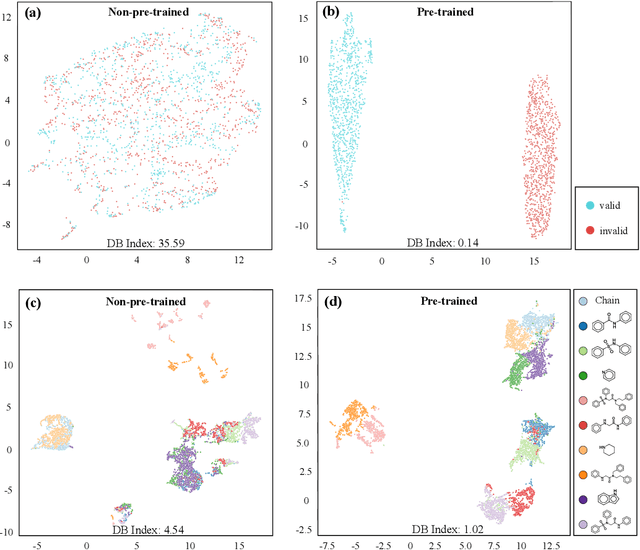

How to produce expressive molecular representations is a fundamental challenge in AI-driven drug discovery. Graph neural network (GNN) has emerged as a powerful technique for modeling molecular data. However, previous supervised approaches usually suffer from the scarcity of labeled data and have poor generalization capability. Here, we proposed a novel Molecular Pre-training Graph-based deep learning framework, named MPG, that leans molecular representations from large-scale unlabeled molecules. In MPG, we proposed a powerful MolGNet model and an effective self-supervised strategy for pre-training the model at both the node and graph-level. After pre-training on 11 million unlabeled molecules, we revealed that MolGNet can capture valuable chemistry insights to produce interpretable representation. The pre-trained MolGNet can be fine-tuned with just one additional output layer to create state-of-the-art models for a wide range of drug discovery tasks, including molecular properties prediction, drug-drug interaction, and drug-target interaction, involving 13 benchmark datasets. Our work demonstrates that MPG is promising to become a novel approach in the drug discovery pipeline.