 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlexible Variational Information Bottleneck: Achieving Diverse Compression with a Single Training
Feb 02, 2024



Information Bottleneck (IB) is a widely used framework that enables the extraction of information related to a target random variable from a source random variable. In the objective function, IB controls the trade-off between data compression and predictiveness through the Lagrange multiplier $\beta$. Traditionally, to find the trade-off to be learned, IB requires a search for $\beta$ through multiple training cycles, which is computationally expensive. In this study, we introduce Flexible Variational Information Bottleneck (FVIB), an innovative framework for classification task that can obtain optimal models for all values of $\beta$ with single, computationally efficient training. We theoretically demonstrate that across all values of reasonable $\beta$, FVIB can simultaneously maximize an approximation of the objective function for Variational Information Bottleneck (VIB), the conventional IB method. Then we empirically show that FVIB can learn the VIB objective as effectively as VIB. Furthermore, in terms of calibration performance, FVIB outperforms other IB and calibration methods by enabling continuous optimization of $\beta$. Our codes are available at https://github.com/sotakudo/fvib.
Pre-training of Molecular GNNs as Conditional Boltzmann Generator
Dec 31, 2023Learning representations of molecular structures using deep learning is a fundamental problem in molecular property prediction tasks. Molecules inherently exist in the real world as three-dimensional structures; furthermore, they are not static but in continuous motion in the 3D Euclidean space, forming a potential energy surface. Therefore, it is desirable to generate multiple conformations in advance and extract molecular representations using a 4D-QSAR model that incorporates multiple conformations. However, this approach is impractical for drug and material discovery tasks because of the computational cost of obtaining multiple conformations. To address this issue, we propose a pre-training method for molecular GNNs using an existing dataset of molecular conformations to generate a latent vector universal to multiple conformations from a 2D molecular graph. Our method, called Boltzmann GNN, is formulated by maximizing the conditional marginal likelihood of a conditional generative model for conformations generation. We show that our model has a better prediction performance for molecular properties than existing pre-training methods using molecular graphs and three-dimensional molecular structures.
Variational Autoencoding Molecular Graphs with Denoising Diffusion Probabilistic Model
Jul 02, 2023In data-driven drug discovery, designing molecular descriptors is a very important task. Deep generative models such as variational autoencoders (VAEs) offer a potential solution by designing descriptors as probabilistic latent vectors derived from molecular structures. These models can be trained on large datasets, which have only molecular structures, and applied to transfer learning. Nevertheless, the approximate posterior distribution of the latent vectors of the usual VAE assumes a simple multivariate Gaussian distribution with zero covariance, which may limit the performance of representing the latent features. To overcome this limitation, we propose a novel molecular deep generative model that incorporates a hierarchical structure into the probabilistic latent vectors. We achieve this by a denoising diffusion probabilistic model (DDPM). We demonstrate that our model can design effective molecular latent vectors for molecular property prediction from some experiments by small datasets on physical properties and activity. The results highlight the superior prediction performance and robustness of our model compared to existing approaches.
Cancer Subtyping by Improved Transcriptomic Features Using Vector Quantized Variational Autoencoder
Jul 20, 2022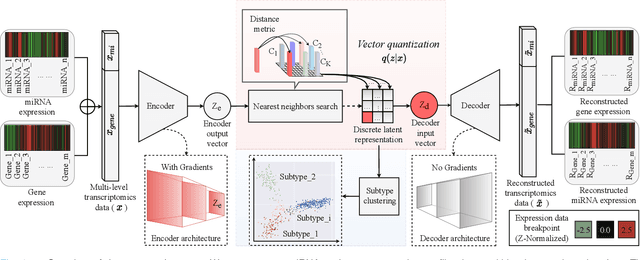
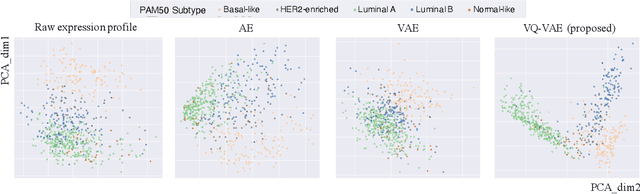
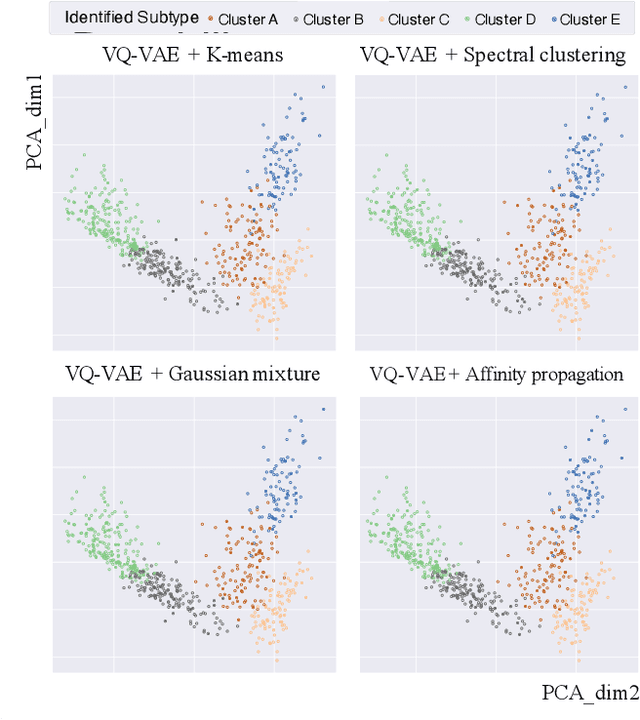
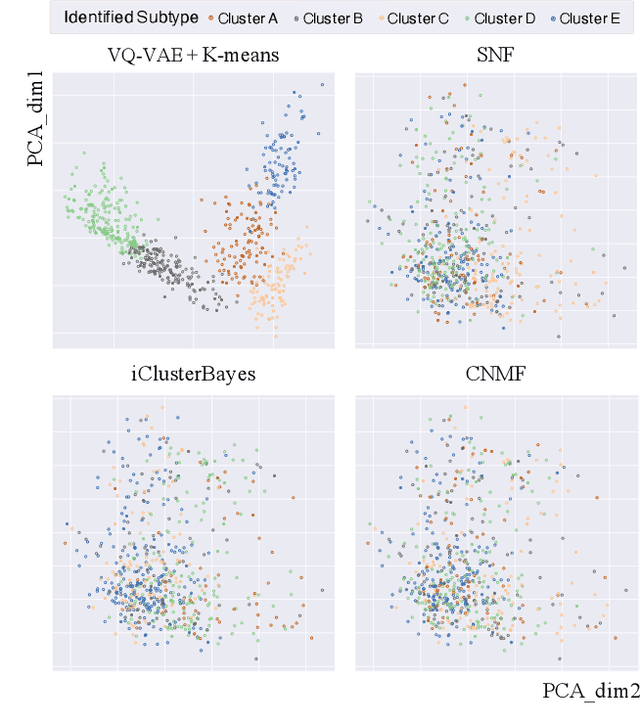
Defining and separating cancer subtypes is essential for facilitating personalized therapy modality and prognosis of patients. The definition of subtypes has been constantly recalibrated as a result of our deepened understanding. During this recalibration, researchers often rely on clustering of cancer data to provide an intuitive visual reference that could reveal the intrinsic characteristics of subtypes. The data being clustered are often omics data such as transcriptomics that have strong correlations to the underlying biological mechanism. However, while existing studies have shown promising results, they suffer from issues associated with omics data: sample scarcity and high dimensionality. As such, existing methods often impose unrealistic assumptions to extract useful features from the data while avoiding overfitting to spurious correlations. In this paper, we propose to leverage a recent strong generative model, Vector Quantized Variational AutoEncoder (VQ-VAE), to tackle the data issues and extract informative latent features that are crucial to the quality of subsequent clustering by retaining only information relevant to reconstructing the input. VQ-VAE does not impose strict assumptions and hence its latent features are better representations of the input, capable of yielding superior clustering performance with any mainstream clustering method. Extensive experiments and medical analysis on multiple datasets comprising 10 distinct cancers demonstrate the VQ-VAE clustering results can significantly and robustly improve prognosis over prevalent subtyping systems.
Enhancement on Model Interpretability and Sleep Stage Scoring Performance with A Novel Pipeline Based on Deep Neural Network
Apr 07, 2022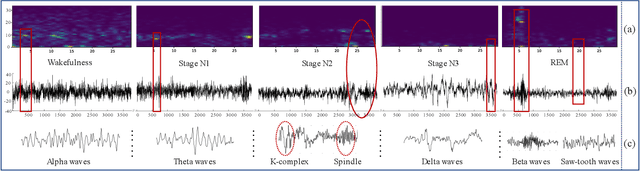
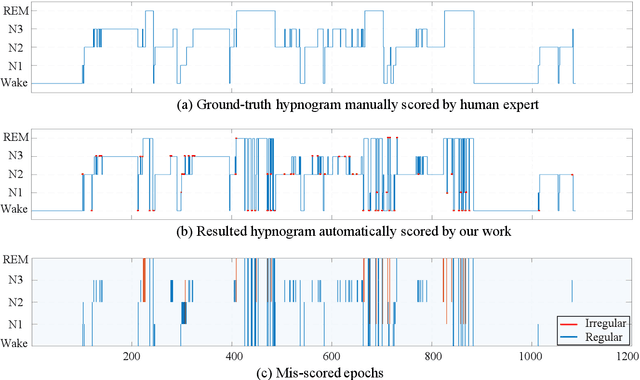

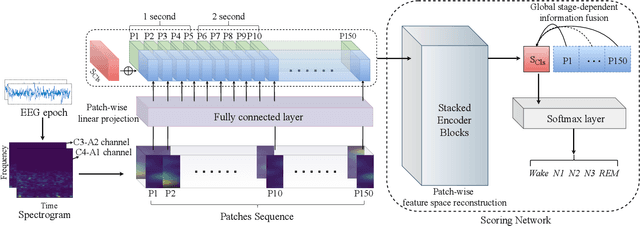
Considering the natural frequency characteristics in sleep medicine, this paper first proposes a time-frequency framework for the representation learning of the electroencephalogram (EEG) following the definition of the American Academy of Sleep Medicine. To meet the temporal-random and transient nature of the defining characteristics of sleep stages, we further design a context-sensitive flexible pipeline that automatically adapts to the attributes of data itself. That is, the input EEG spectrogram is partitioned into a sequence of patches in the time and frequency axes, and then input to a delicate deep learning network for further representation learning to extract the stage-dependent features, which are used in the classification step finally. The proposed pipeline is validated against a large database, i.e., the Sleep Heart Health Study (SHHS), and the results demonstrate that the competitive performance for the wake, N2, and N3 stages outperforms the state-of-art works, with the F1 scores being 0.93, 0.88, and 0.87, respectively, and the proposed method has a high inter-rater reliability of 0.80 kappa. Importantly, we visualize the stage scoring process of the model decision with the Layer-wise Relevance Propagation (LRP) method, which shows that the proposed pipeline is more sensitive and perceivable in the decision-making process than the baseline pipelines. Therefore, the pipeline together with the LRP method can provide better model interpretability, which is important for clinical support.
Cancer Subtyping via Embedded Unsupervised Learning on Transcriptomics Data
Apr 02, 2022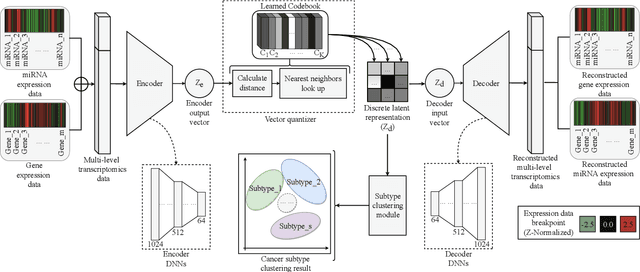
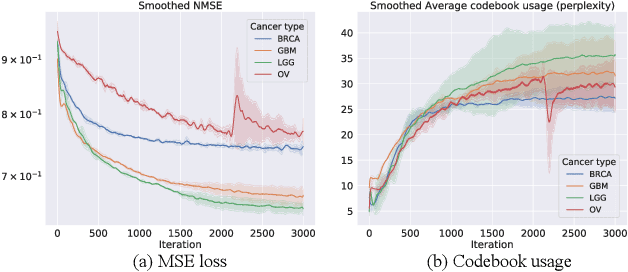
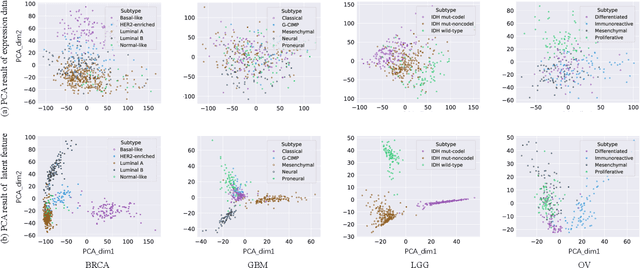

Cancer is one of the deadliest diseases worldwide. Accurate diagnosis and classification of cancer subtypes are indispensable for effective clinical treatment. Promising results on automatic cancer subtyping systems have been published recently with the emergence of various deep learning methods. However, such automatic systems often overfit the data due to the high dimensionality and scarcity. In this paper, we propose to investigate automatic subtyping from an unsupervised learning perspective by directly constructing the underlying data distribution itself, hence sufficient data can be generated to alleviate the issue of overfitting. Specifically, we bypass the strong Gaussianity assumption that typically exists but fails in the unsupervised learning subtyping literature due to small-sized samples by vector quantization. Our proposed method better captures the latent space features and models the cancer subtype manifestation on a molecular basis, as demonstrated by the extensive experimental results.