 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe IBEX Imaging Knowledge-Base: A Community Resource Enabling Adoption and Development of Immunofluoresence Imaging Methods
Dec 17, 2024The iterative bleaching extends multiplexity (IBEX) Knowledge-Base is a central portal for researchers adopting IBEX and related 2D and 3D immunofluorescence imaging methods. The design of the Knowledge-Base is modeled after efforts in the open-source software community and includes three facets: a development platform (GitHub), static website, and service for data archiving. The Knowledge-Base facilitates the practice of open science throughout the research life cycle by providing validation data for recommended and non-recommended reagents, e.g., primary and secondary antibodies. In addition to reporting negative data, the Knowledge-Base empowers method adoption and evolution by providing a venue for sharing protocols, videos, datasets, software, and publications. A dedicated discussion forum fosters a sense of community among researchers while addressing questions not covered in published manuscripts. Together, scientists from around the world are advancing scientific discovery at a faster pace, reducing wasted time and effort, and instilling greater confidence in the resulting data.
Celcomen: spatial causal disentanglement for single-cell and tissue perturbation modeling
Sep 09, 2024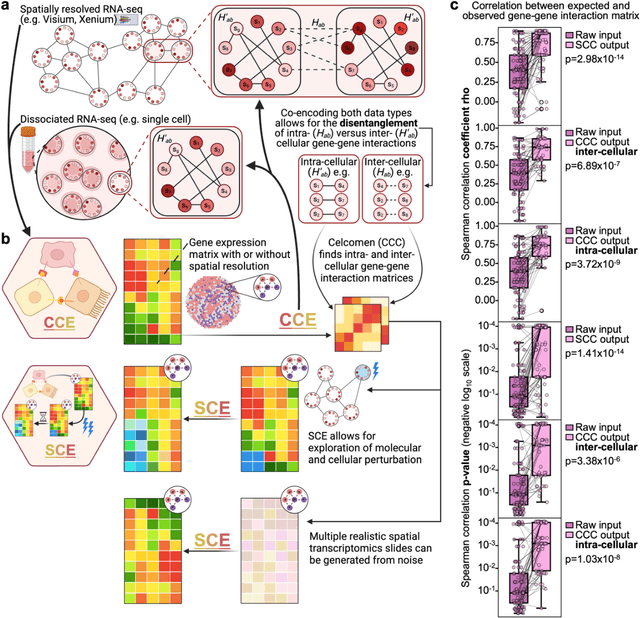
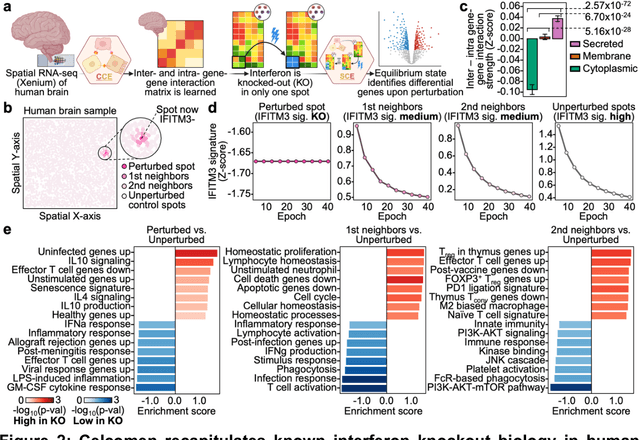
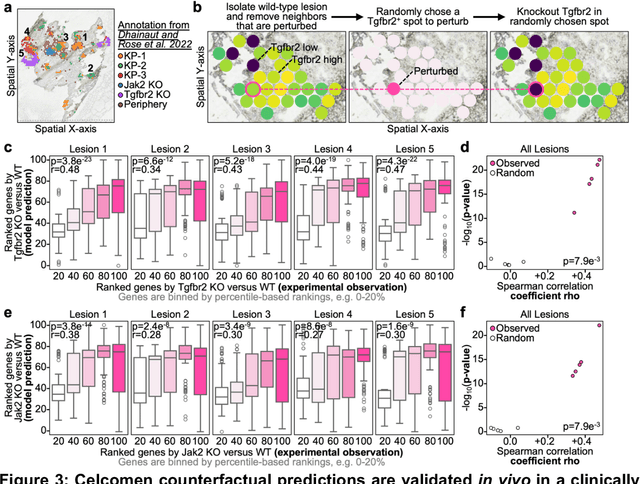
Celcomen leverages a mathematical causality framework to disentangle intra- and inter- cellular gene regulation programs in spatial transcriptomics and single-cell data through a generative graph neural network. It can learn gene-gene interactions, as well as generate post-perturbation counterfactual spatial transcriptomics, thereby offering access to experimentally inaccessible samples. We validated its disentanglement, identifiability, and counterfactual prediction capabilities through simulations and in clinically relevant human glioblastoma, human fetal spleen, and mouse lung cancer samples. Celcomen provides the means to model disease and therapy induced changes allowing for new insights into single-cell spatially resolved tissue responses relevant to human health.
Modelling Technical and Biological Effects in scRNA-seq data with Scalable GPLVMs
Sep 14, 2022



Single-cell RNA-seq datasets are growing in size and complexity, enabling the study of cellular composition changes in various biological/clinical contexts. Scalable dimensionality reduction techniques are in need to disentangle biological variation in them, while accounting for technical and biological confounders. In this work, we extend a popular approach for probabilistic non-linear dimensionality reduction, the Gaussian process latent variable model, to scale to massive single-cell datasets while explicitly accounting for technical and biological confounders. The key idea is to use an augmented kernel which preserves the factorisability of the lower bound allowing for fast stochastic variational inference. We demonstrate its ability to reconstruct latent signatures of innate immunity recovered in Kumasaka et al. (2021) with 9x lower training time. We further analyze a COVID dataset and demonstrate across a cohort of 130 individuals, that this framework enables data integration while capturing interpretable signatures of infection. Specifically, we explore COVID severity as a latent dimension to refine patient stratification and capture disease-specific gene expression.