 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA comparative study of similarity-based and GNN-based link prediction approaches
Aug 20, 2020


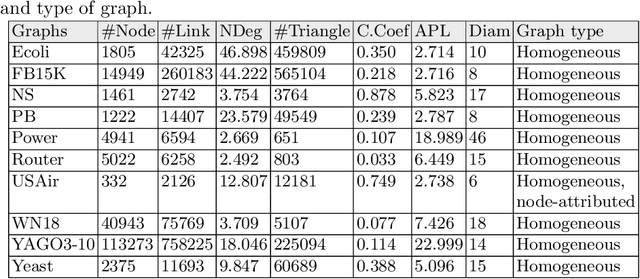
The task of inferring the missing links in a graph based on its current structure is referred to as link prediction. Link prediction methods that are based on pairwise node similarity are well-established approaches in the literature. They show good prediction performance in many real-world graphs though they are heuristics and lack of universal applicability. On the other hand, the success of neural networks for classification tasks in various domains leads researchers to study them in graphs. When a neural network can operate directly on the graph, then it is termed as the graph neural network (GNN). GNN is able to learn hidden features from graphs which can be used for link prediction task in graphs. Link predictions based on GNNs have gained much attention of researchers due to their convincing high performance in many real-world graphs. This appraisal paper studies some similarity and GNN-based link prediction approaches in the domain of homogeneous graphs that consists of a single type of (attributed) nodes and single type of pairwise links. We evaluate the studied approaches against several benchmark graphs with different properties from various domains.
Neighborhood-Based Label Propagation in Large Protein Graphs
Aug 09, 2017
Understanding protein function is one of the keys to understanding life at the molecular level. It is also important in several scenarios including human disease and drug discovery. In this age of rapid and affordable biological sequencing, the number of sequences accumulating in databases is rising with an increasing rate. This presents many challenges for biologists and computer scientists alike. In order to make sense of this huge quantity of data, these sequences should be annotated with functional properties. UniProtKB consists of two components: i) the UniProtKB/Swiss-Prot database containing protein sequences with reliable information manually reviewed by expert bio-curators and ii) the UniProtKB/TrEMBL database that is used for storing and processing the unknown sequences. Hence, for all proteins we have available the sequence along with few more information such as the taxon and some structural domains. Pairwise similarity can be defined and computed on proteins based on such attributes. Other important attributes, while present for proteins in Swiss-Prot, are often missing for proteins in TrEMBL, such as their function and cellular localization. The enormous number of protein sequences now in TrEMBL calls for rapid procedures to annotate them automatically. In this work, we present DistNBLP, a novel Distributed Neighborhood-Based Label Propagation approach for large-scale annotation of proteins. To do this, the functional annotations of reviewed proteins are used to predict those of non-reviewed proteins using label propagation on a graph representation of the protein database. DistNBLP is built on top of the "akka" toolkit for building resilient distributed message-driven applications.
Scalable Semi-Supervised Learning over Networks using Nonsmooth Convex Optimization
Nov 02, 2016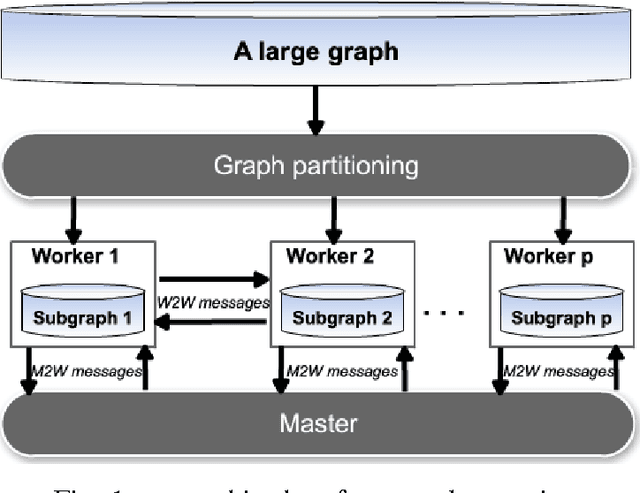
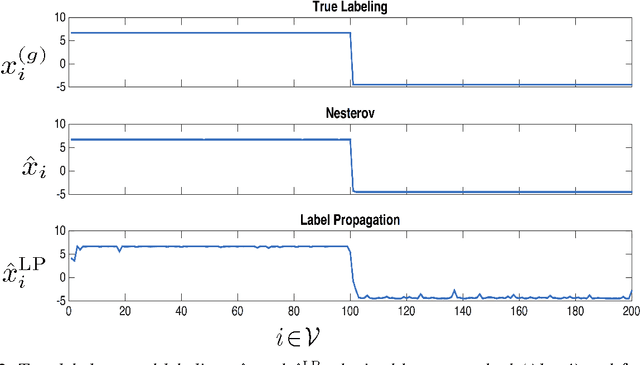
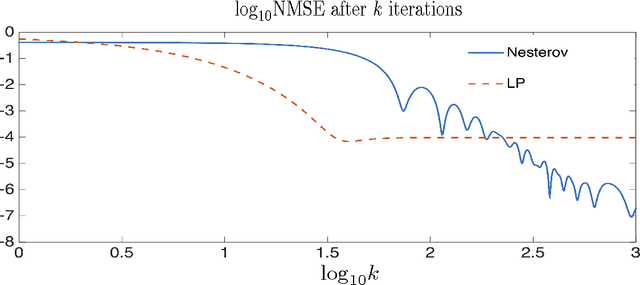
We propose a scalable method for semi-supervised (transductive) learning from massive network-structured datasets. Our approach to semi-supervised learning is based on representing the underlying hypothesis as a graph signal with small total variation. Requiring a small total variation of the graph signal representing the underlying hypothesis corresponds to the central smoothness assumption that forms the basis for semi-supervised learning, i.e., input points forming clusters have similar output values or labels. We formulate the learning problem as a nonsmooth convex optimization problem which we solve by appealing to Nesterovs optimal first-order method for nonsmooth optimization. We also provide a message passing formulation of the learning method which allows for a highly scalable implementation in big data frameworks.
A multiple instance learning approach for sequence data with across bag dependencies
Jan 30, 2016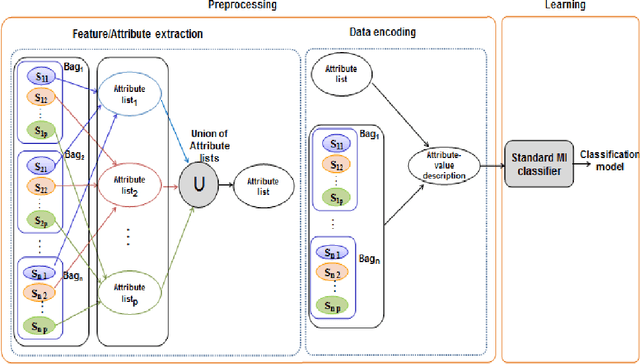
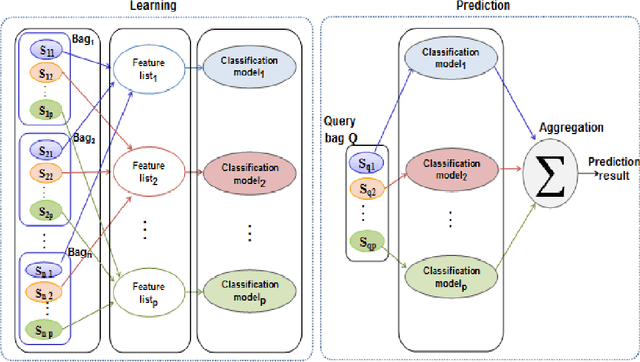
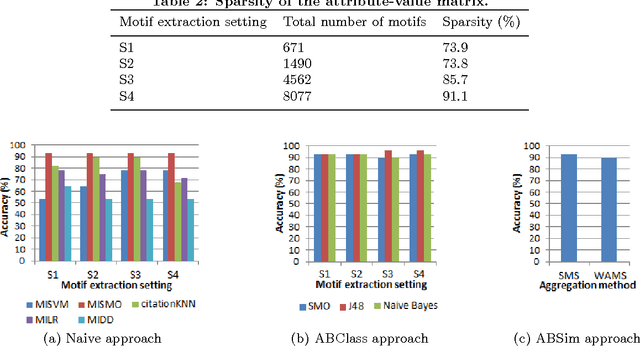
In Multiple Instance Learning (MIL) problem for sequence data, the learning data consist of a set of bags where each bag contains a set of instances/sequences. In many real world applications such as bioinformatics, web mining, and text mining, comparing a random couple of sequences makes no sense. In fact, each instance of each bag may have structural and/or temporal relation with other instances in other bags. Thus, the classification task should take into account the relation between semantically related instances across bags. In this paper, we present two novel MIL approaches for sequence data classification: (1) ABClass and (2) ABSim. In ABClass, each sequence is represented by one vector of attributes. For each sequence of the unknown bag, a discriminative classifier is applied in order to compute a partial classification result. Then, an aggregation method is applied to these partial results in order to generate the final result. In ABSim, we use a similarity measure between each sequence of the unknown bag and the corresponding sequences in the learning bags. An unknown bag is labeled with the bag that presents more similar sequences. We applied both approaches to the problem of bacterial Ionizing Radiation Resistance (IRR) prediction. We evaluated and discussed the proposed approaches on well known Ionizing Radiation Resistance Bacteria (IRRB) and Ionizing Radiation Sensitive Bacteria (IRSB) represented by primary structure of basal DNA repair proteins. The experimental results show that both ABClass and ABSim approaches are efficient.
Towards a constructive multilayer perceptron for regression task using non-parametric clustering. A case study of Photo-Z redshift reconstruction
Dec 17, 2014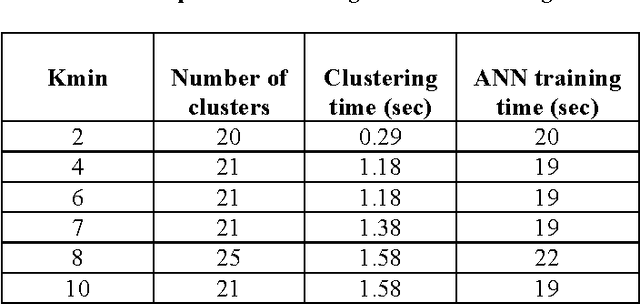
The choice of architecture of artificial neuron network (ANN) is still a challenging task that users face every time. It greatly affects the accuracy of the built network. In fact there is no optimal method that is applicable to various implementations at the same time. In this paper we propose a method to construct ANN based on clustering, that resolves the problems of random and ad hoc approaches for multilayer ANN architecture. Our method can be applied to regression problems. Experimental results obtained with different datasets, reveals the efficiency of our method.
Feature extraction in protein sequences classification : a new stability measure
Dec 05, 2012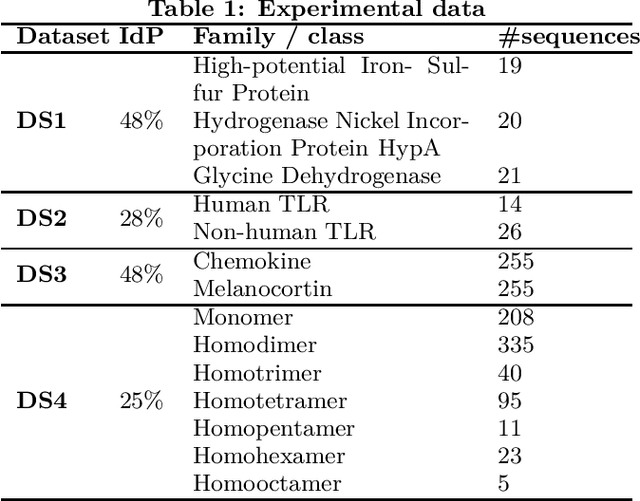
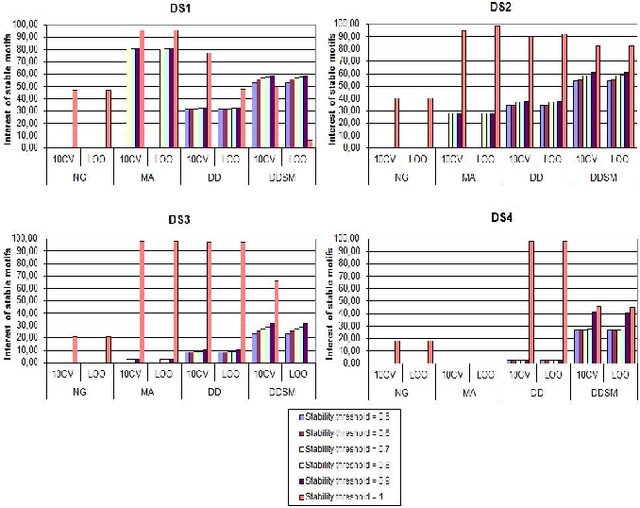
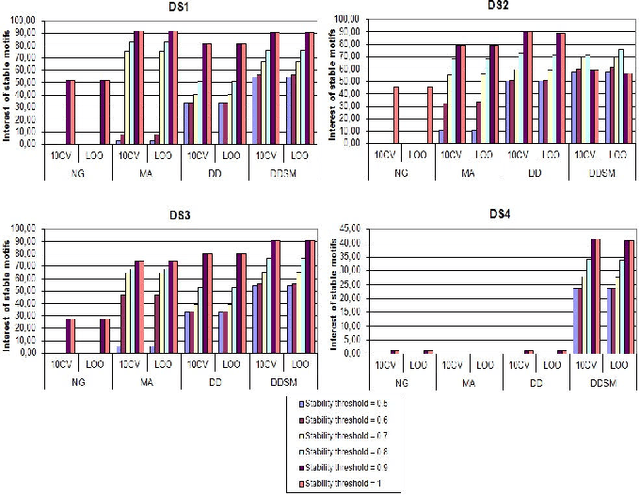
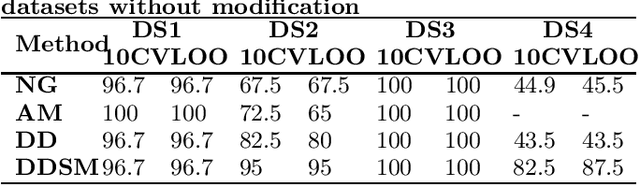
Feature extraction is an unavoidable task, especially in the critical step of preprocessing biological sequences. This step consists for example in transforming the biological sequences into vectors of motifs where each motif is a subsequence that can be seen as a property (or attribute) characterizing the sequence. Hence, we obtain an object-property table where objects are sequences and properties are motifs extracted from sequences. This output can be used to apply standard machine learning tools to perform data mining tasks such as classification. Several previous works have described feature extraction methods for bio-sequence classification, but none of them discussed the robustness of these methods when perturbing the input data. In this work, we introduce the notion of stability of the generated motifs in order to study the robustness of motif extraction methods. We express this robustness in terms of the ability of the method to reveal any change occurring in the input data and also its ability to target the interesting motifs. We use these criteria to evaluate and experimentally compare four existing extraction methods for biological sequences.