 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMining Representative Unsubstituted Graph Patterns Using Prior Similarity Matrix
Mar 08, 2013



One of the most powerful techniques to study protein structures is to look for recurrent fragments (also called substructures or spatial motifs), then use them as patterns to characterize the proteins under study. An emergent trend consists in parsing proteins three-dimensional (3D) structures into graphs of amino acids. Hence, the search of recurrent spatial motifs is formulated as a process of frequent subgraph discovery where each subgraph represents a spatial motif. In this scope, several efficient approaches for frequent subgraph discovery have been proposed in the literature. However, the set of discovered frequent subgraphs is too large to be efficiently analyzed and explored in any further process. In this paper, we propose a novel pattern selection approach that shrinks the large number of discovered frequent subgraphs by selecting the representative ones. Existing pattern selection approaches do not exploit the domain knowledge. Yet, in our approach we incorporate the evolutionary information of amino acids defined in the substitution matrices in order to select the representative subgraphs. We show the effectiveness of our approach on a number of real datasets. The results issued from our experiments show that our approach is able to considerably decrease the number of motifs while enhancing their interestingness.
Feature extraction in protein sequences classification : a new stability measure
Dec 05, 2012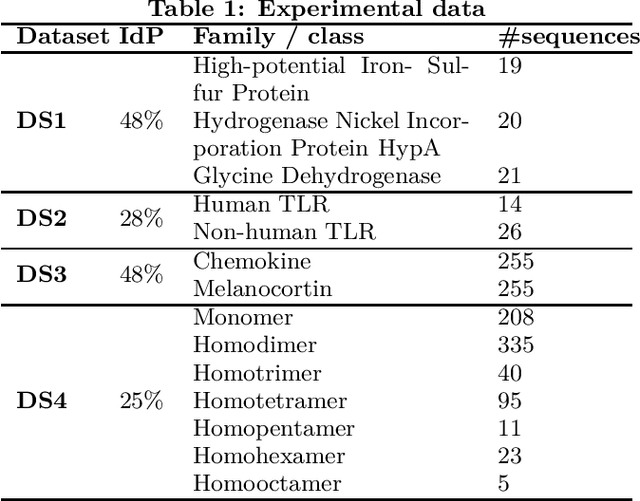
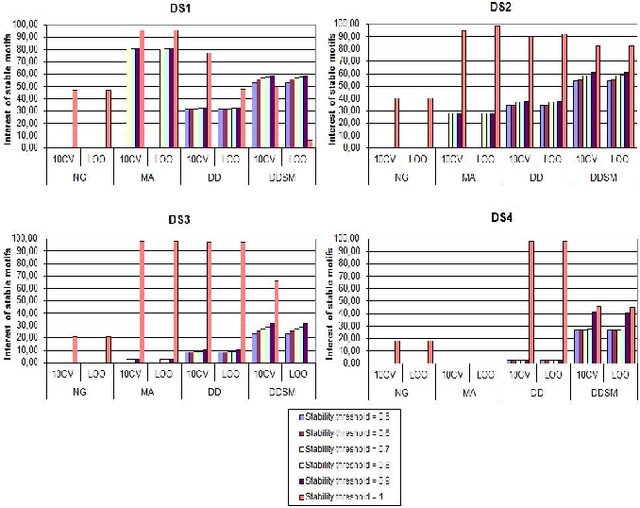
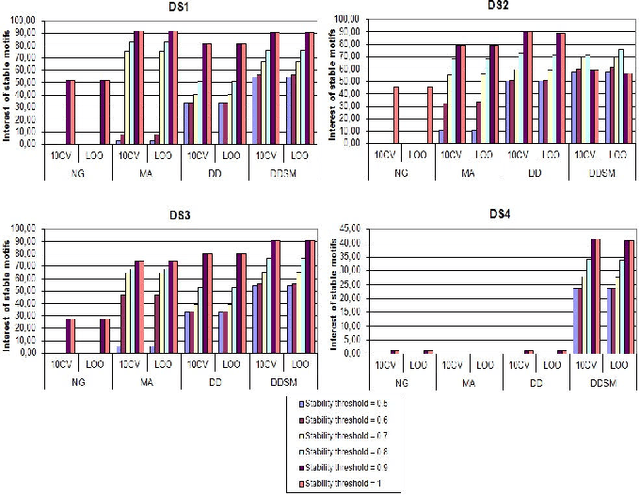
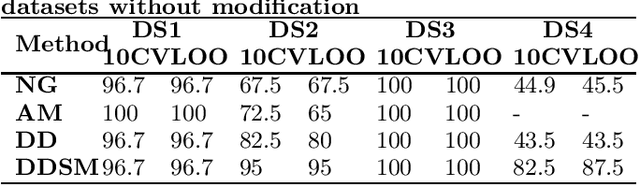
Feature extraction is an unavoidable task, especially in the critical step of preprocessing biological sequences. This step consists for example in transforming the biological sequences into vectors of motifs where each motif is a subsequence that can be seen as a property (or attribute) characterizing the sequence. Hence, we obtain an object-property table where objects are sequences and properties are motifs extracted from sequences. This output can be used to apply standard machine learning tools to perform data mining tasks such as classification. Several previous works have described feature extraction methods for bio-sequence classification, but none of them discussed the robustness of these methods when perturbing the input data. In this work, we introduce the notion of stability of the generated motifs in order to study the robustness of motif extraction methods. We express this robustness in terms of the ability of the method to reveal any change occurring in the input data and also its ability to target the interesting motifs. We use these criteria to evaluate and experimentally compare four existing extraction methods for biological sequences.