 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom SIR to SEAIRD: a novel data-driven modeling approach based on the Grey-box System Theory to predict the dynamics of COVID-19
May 29, 2021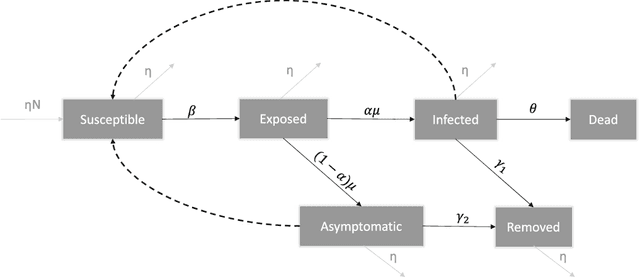
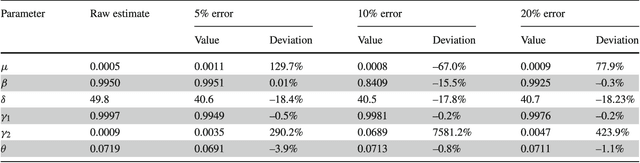
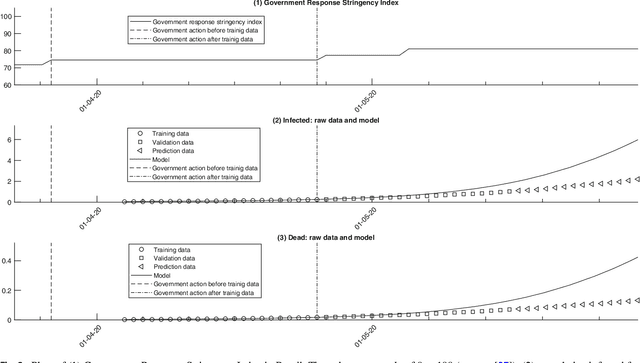
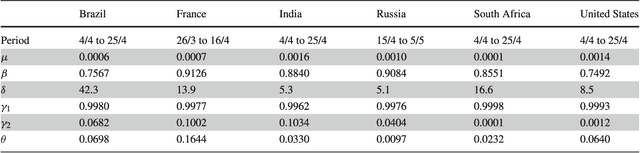
Common compartmental modeling for COVID-19 is based on a priori knowledge and numerous assumptions. Additionally, they do not systematically incorporate asymptomatic cases. Our study aimed at providing a framework for data-driven approaches, by leveraging the strengths of the grey-box system theory or grey-box identification, known for its robustness in problem solving under partial, incomplete, or uncertain data. Empirical data on confirmed cases and deaths, extracted from an open source repository were used to develop the SEAIRD compartment model. Adjustments were made to fit current knowledge on the COVID-19 behavior. The model was implemented and solved using an Ordinary Differential Equation solver and an optimization tool. A cross-validation technique was applied, and the coefficient of determination $R^2$ was computed in order to evaluate the goodness-of-fit of the model. %to the data. Key epidemiological parameters were finally estimated and we provided the rationale for the construction of SEAIRD model. When applied to Brazil's cases, SEAIRD produced an excellent agreement to the data, with an %coefficient of determination $R^2$ $\geq 90\%$. The probability of COVID-19 transmission was generally high ($\geq 95\%$). On the basis of a 20-day modeling data, the incidence rate of COVID-19 was as low as 3 infected cases per 100,000 exposed persons in Brazil and France. Within the same time frame, the fatality rate of COVID-19 was the highest in France (16.4\%) followed by Brazil (6.9\%), and the lowest in Russia ($\leq 1\%$). SEAIRD represents an asset for modeling infectious diseases in their dynamical stable phase, especially for new viruses when pathophysiology knowledge is very limited.
Toward an Efficient Multi-class Classification in an Open Universe
Mar 01, 2018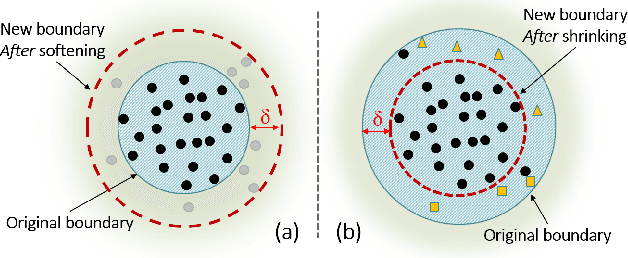
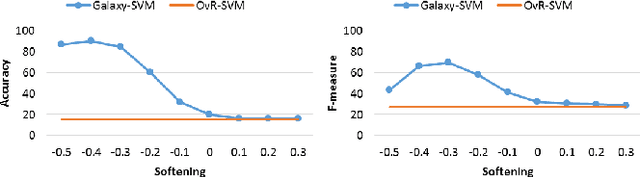

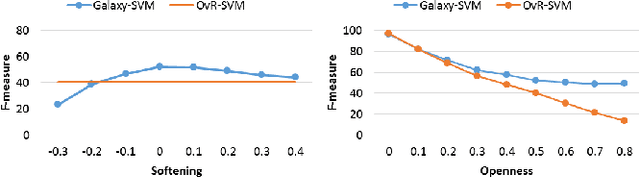
Classification is a fundamental task in machine learning and data mining. Existing classification methods are designed to classify unknown instances within a set of previously known training classes. Such a classification takes the form of a prediction within a closed-set of classes. However, a more realistic scenario that fits real-world applications is to consider the possibility of encountering instances that do not belong to any of the training classes, $i.e.$, an open-set classification. In such situation, existing closed-set classifiers will assign a training label to these instances resulting in a misclassification. In this paper, we introduce Galaxy-X, a novel multi-class classification approach for open-set recognition problems. For each class of the training set, Galaxy-X creates a minimum bounding hyper-sphere that encompasses the distribution of the class by enclosing all of its instances. In such manner, our method is able to distinguish instances resembling previously seen classes from those that are of unknown ones. To adequately evaluate open-set classification, we introduce a novel evaluation procedure. Experimental results on benchmark datasets show the efficiency of our approach in classifying novel instances from known as well as unknown classes.
A large scale study of SVM based methods for abstract screening in systematic reviews
Jan 16, 2018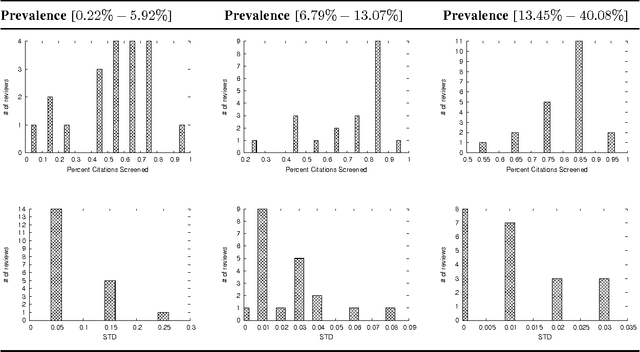
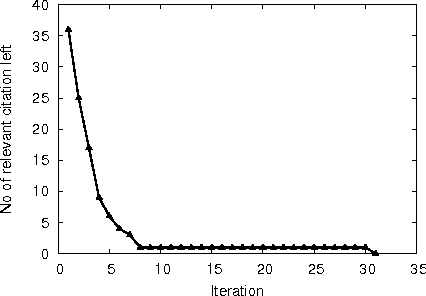


A major task in systematic reviews is abstract screening, i.e., excluding, often hundreds or thousand of, irrelevant citations returned from a database search based on titles and abstracts. Thus, a systematic review platform that can automate the abstract screening process is of huge importance. Several methods have been proposed for this task. However, it is very hard to clearly understand the applicability of these methods in a systematic review platform because of the following challenges: (1) the use of non-overlapping metrics for the evaluation of the proposed methods, (2) usage of features that are very hard to collect, (3) using a small set of reviews for the evaluation, and (4) no solid statistical testing or equivalence grouping of the methods. In this paper, we use feature representation that can be extracted per citation. We evaluate SVM-based methods (commonly used) on a large set of reviews ($61$) and metrics ($11$) to provide equivalence grouping of methods based on a solid statistical test. Our analysis also includes a strong variability of the metrics using $500$x$2$ cross validation. While some methods shine for different metrics and for different datasets, there is no single method that dominates the pack. Furthermore, we observe that in some cases relevant (included) citations can be found after screening only 15-20% of them via a certainty based sampling. A few included citations present outlying characteristics and can only be found after a very large number of screening steps. Finally, we present an ensemble algorithm for producing a $5$-star rating of citations based on their relevance. Such algorithm combines the best methods from our evaluation and through its $5$-star rating outputs a more easy-to-consume prediction.
ProtNN: Fast and Accurate Nearest Neighbor Protein Function Prediction based on Graph Embedding in Structural and Topological Space
Jan 25, 2016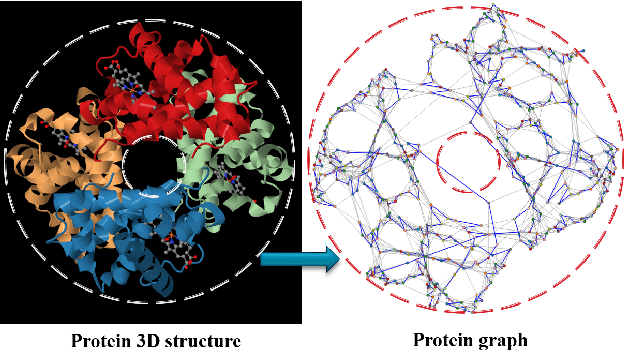
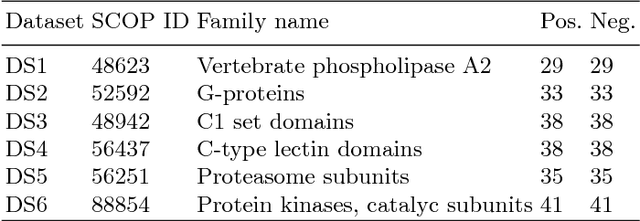
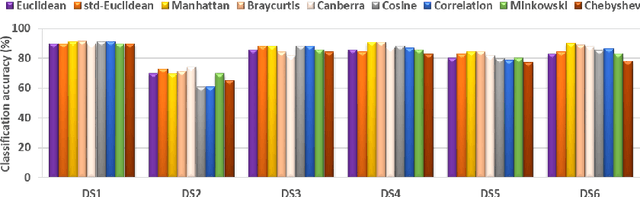

Studying the function of proteins is important for understanding the molecular mechanisms of life. The number of publicly available protein structures has increasingly become extremely large. Still, the determination of the function of a protein structure remains a difficult, costly, and time consuming task. The difficulties are often due to the essential role of spatial and topological structures in the determination of protein functions in living cells. In this paper, we propose ProtNN, a novel approach for protein function prediction. Given an unannotated protein structure and a set of annotated proteins, ProtNN finds the nearest neighbor annotated structures based on protein-graph pairwise similarities. Given a query protein, ProtNN finds the nearest neighbor reference proteins based on a graph representation model and a pairwise similarity between vector embedding of both query and reference protein-graphs in structural and topological spaces. ProtNN assigns to the query protein the function with the highest number of votes across the set of k nearest neighbor reference proteins, where k is a user-defined parameter. Experimental evaluation demonstrates that ProtNN is able to accurately classify several datasets in an extremely fast runtime compared to state-of-the-art approaches. We further show that ProtNN is able to scale up to a whole PDB dataset in a single-process mode with no parallelization, with a gain of thousands order of magnitude of runtime compared to state-of-the-art approaches.
Mining Representative Unsubstituted Graph Patterns Using Prior Similarity Matrix
Mar 08, 2013



One of the most powerful techniques to study protein structures is to look for recurrent fragments (also called substructures or spatial motifs), then use them as patterns to characterize the proteins under study. An emergent trend consists in parsing proteins three-dimensional (3D) structures into graphs of amino acids. Hence, the search of recurrent spatial motifs is formulated as a process of frequent subgraph discovery where each subgraph represents a spatial motif. In this scope, several efficient approaches for frequent subgraph discovery have been proposed in the literature. However, the set of discovered frequent subgraphs is too large to be efficiently analyzed and explored in any further process. In this paper, we propose a novel pattern selection approach that shrinks the large number of discovered frequent subgraphs by selecting the representative ones. Existing pattern selection approaches do not exploit the domain knowledge. Yet, in our approach we incorporate the evolutionary information of amino acids defined in the substitution matrices in order to select the representative subgraphs. We show the effectiveness of our approach on a number of real datasets. The results issued from our experiments show that our approach is able to considerably decrease the number of motifs while enhancing their interestingness.