 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Unified Perspective for Learning Graph Representations Across Multi-Level Abstractions
May 12, 2026Graph Self-Supervised Learning (GSSL) has emerged as a powerful paradigm for generating high-quality representations for graph-structured data. While multi-scale graph contrastive learning has received increasing attention, many existing methods still predominantly focus on a single graph abstraction level. To address this limitation, we propose a unified contrastive framework that can target node-level, proximity-level, cluster-level, and graph-level information and integrate them through a linear combination of similarity scores on positive pairs and dissimilarity scores (i.e., similarity scores on negative pairs). Furthermore, current approaches typically assign uniform penalty strengths to all examples, which reduces optimization flexibility and leads to ambiguous convergence status. To overcome this, we introduce a novel parameter-free fine-grained self-weighting mechanism that adaptively assigns weights to individual similarity and dissimilarity scores. The proposed mechanism emphasizes the scores that deviate significantly from their target values. Our approach not only enhances optimization flexibility but also eliminates the computational overhead of hyperparameter tuning in conventional multi-task GSSL methods. Comprehensive experiments on real-world datasets show that our methods consistently outperform state-of-the-art approaches across downstream tasks, including classification, clustering, and link prediction, in both single-level and multi-level scenarios.
A Green Multi-Attribute Client Selection for Over-The-Air Federated Learning: A Grey-Wolf-Optimizer Approach
Sep 16, 2024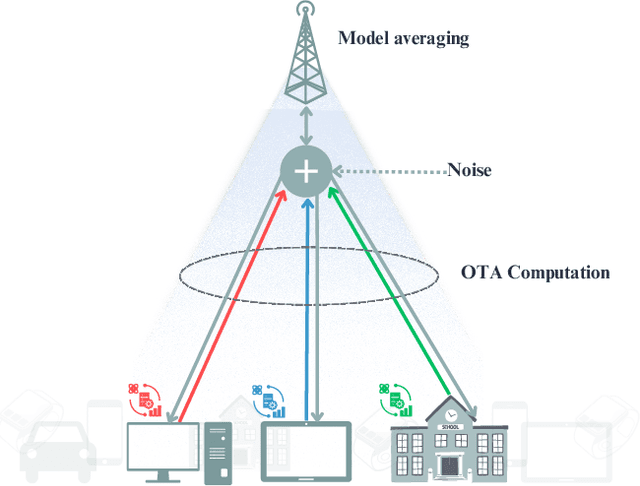
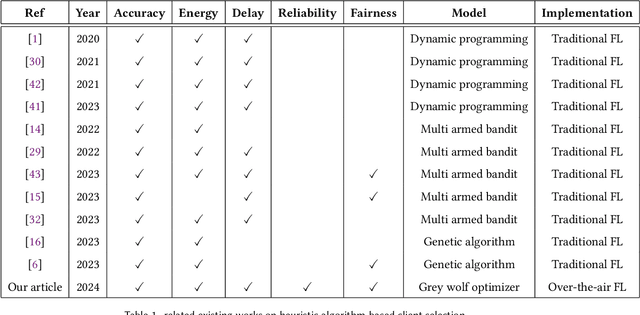

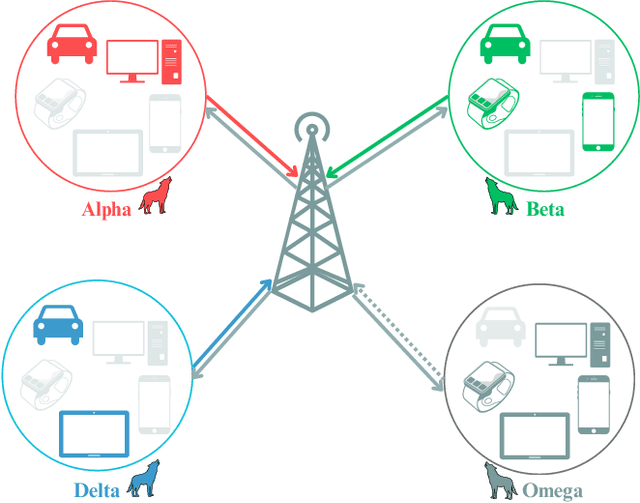
Federated Learning (FL) has gained attention across various industries for its capability to train machine learning models without centralizing sensitive data. While this approach offers significant benefits such as privacy preservation and decreased communication overhead, it presents several challenges, including deployment complexity and interoperability issues, particularly in heterogeneous scenarios or resource-constrained environments. Over-the-air (OTA) FL was introduced to tackle these challenges by disseminating model updates without necessitating direct device-to-device connections or centralized servers. However, OTA-FL brought forth limitations associated with heightened energy consumption and network latency. In this paper, we propose a multi-attribute client selection framework employing the grey wolf optimizer (GWO) to strategically control the number of participants in each round and optimize the OTA-FL process while considering accuracy, energy, delay, reliability, and fairness constraints of participating devices. We evaluate the performance of our multi-attribute client selection approach in terms of model loss minimization, convergence time reduction, and energy efficiency. In our experimental evaluation, we assessed and compared the performance of our approach against the existing state-of-the-art methods. Our results demonstrate that the proposed GWO-based client selection outperforms these baselines across various metrics. Specifically, our approach achieves a notable reduction in model loss, accelerates convergence time, and enhances energy efficiency while maintaining high fairness and reliability indicators.
The Canadian Cropland Dataset: A New Land Cover Dataset for Multitemporal Deep Learning Classification in Agriculture
Jun 04, 2023



Monitoring land cover using remote sensing is vital for studying environmental changes and ensuring global food security through crop yield forecasting. Specifically, multitemporal remote sensing imagery provides relevant information about the dynamics of a scene, which has proven to lead to better land cover classification results. Nevertheless, few studies have benefited from high spatial and temporal resolution data due to the difficulty of accessing reliable, fine-grained and high-quality annotated samples to support their hypotheses. Therefore, we introduce a temporal patch-based dataset of Canadian croplands, enriched with labels retrieved from the Canadian Annual Crop Inventory. The dataset contains 78,536 manually verified high-resolution (10 m/pixel, 640 x 640 m) geo-referenced images from 10 crop classes collected over four crop production years (2017-2020) and five months (June-October). Each instance contains 12 spectral bands, an RGB image, and additional vegetation index bands. Individually, each category contains at least 4,800 images. Moreover, as a benchmark, we provide models and source code that allow a user to predict the crop class using a single image (ResNet, DenseNet, EfficientNet) or a sequence of images (LRCN, 3D-CNN) from the same location. In perspective, we expect this evolving dataset to propel the creation of robust agro-environmental models that can accelerate the comprehension of complex agricultural regions by providing accurate and continuous monitoring of land cover.
Prior Density Learning in Variational Bayesian Phylogenetic Parameters Inference
Feb 06, 2023The advances in variational inference are providing promising paths in Bayesian estimation problems. These advances make variational phylogenetic inference an alternative approach to Markov Chain Monte Carlo methods for approximating the phylogenetic posterior. However, one of the main drawbacks of such approaches is the modelling of the prior through fixed distributions, which could bias the posterior approximation if they are distant from the current data distribution. In this paper, we propose an approach and an implementation framework to relax the rigidity of the prior densities by learning their parameters using a gradient-based method and a neural network-based parameterization. We applied this approach for branch lengths and evolutionary parameters estimation under several Markov chain substitution models. The results of performed simulations show that the approach is powerful in estimating branch lengths and evolutionary model parameters. They also show that a flexible prior model could provide better results than a predefined prior model. Finally, the results highlight that using neural networks improves the initialization of the optimization of the prior density parameters.
EvoVGM: A Deep Variational Generative Model for Evolutionary Parameter Estimation
May 25, 2022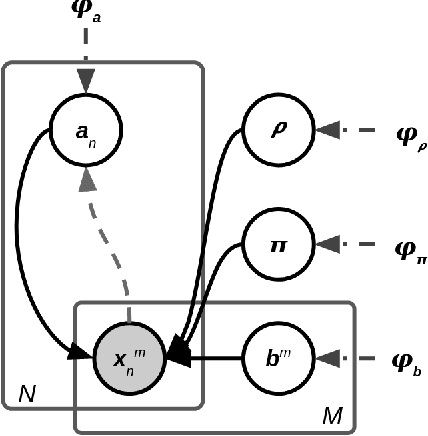
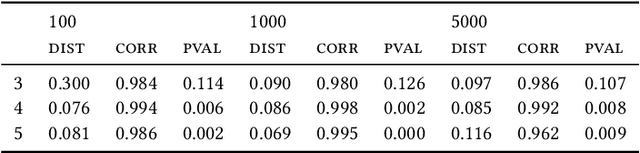
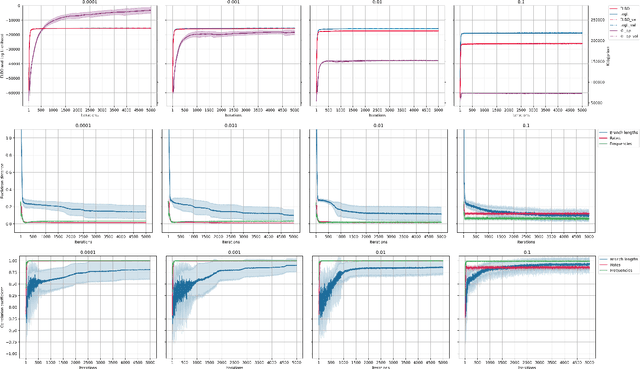
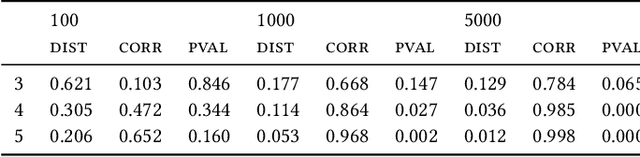
Most evolutionary-oriented deep generative models do not explicitly consider the underlying evolutionary dynamics of biological sequences as it is performed within the Bayesian phylogenetic inference framework. In this study, we propose a method for a deep variational Bayesian generative model that jointly approximates the true posterior of local biological evolutionary parameters and generates sequence alignments. Moreover, it is instantiated and tuned for continuous-time Markov chain substitution models such as JC69 and GTR. We train the model via a low-variance variational objective function and a gradient ascent algorithm. Here, we show the consistency and effectiveness of the method on synthetic sequence alignments simulated with several evolutionary scenarios and on a real virus sequence alignment.
Supporting supervised learning in fungal Biosynthetic Gene Cluster discovery: new benchmark datasets
Jan 09, 2020
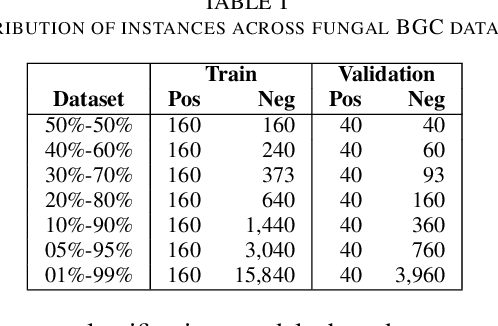
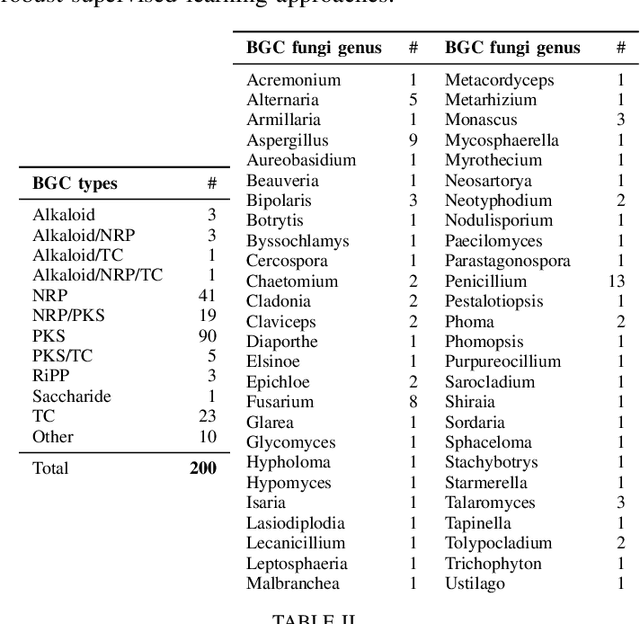

Fungal Biosynthetic Gene Clusters (BGCs) of secondary metabolites are clusters of genes capable of producing natural products, compounds that play an important role in the production of a wide variety of bioactive compounds, including antibiotics and pharmaceuticals. Identifying BGCs can lead to the discovery of novel natural products to benefit human health. Previous work has been focused on developing automatic tools to support BGC discovery in plants, fungi, and bacteria. Data-driven methods, as well as probabilistic and supervised learning methods have been explored in identifying BGCs. Most methods applied to identify fungal BGCs were data-driven and presented limited scope. Supervised learning methods have been shown to perform well at identifying BGCs in bacteria, and could be well suited to perform the same task in fungi. But labeled data instances are needed to perform supervised learning. Openly accessible BGC databases contain only a very small portion of previously curated fungal BGCs. Making new fungal BGC datasets available could motivate the development of supervised learning methods for fungal BGCs and potentially improve prediction performance compared to data-driven methods. In this work we propose new publicly available fungal BGC datasets to support the BGC discovery task using supervised learning. These datasets are prepared to perform binary classification and predict candidate BGC regions in fungal genomes. In addition we analyse the performance of a well supported supervised learning tool developed to predict BGCs.
Statistical Linear Models in Virus Genomic Alignment-free Classification: Application to Hepatitis C Viruses
Nov 06, 2019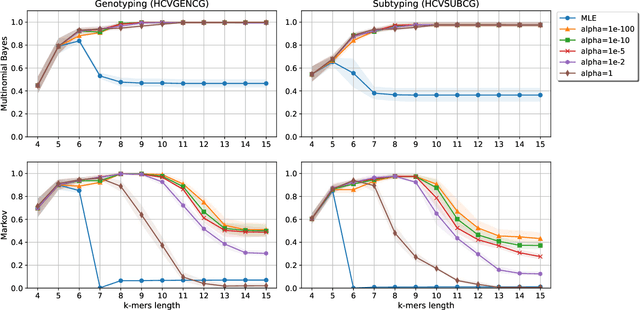
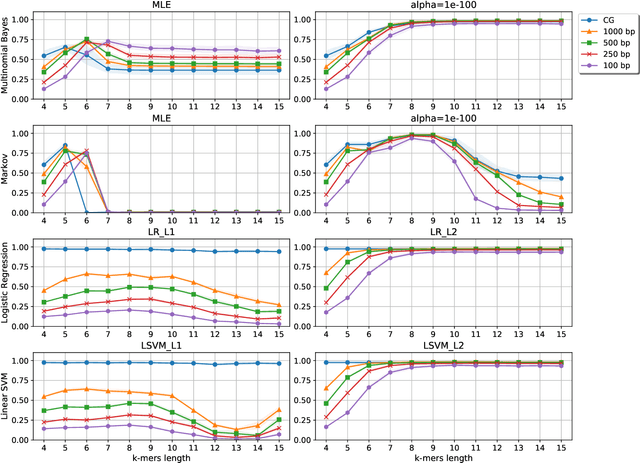
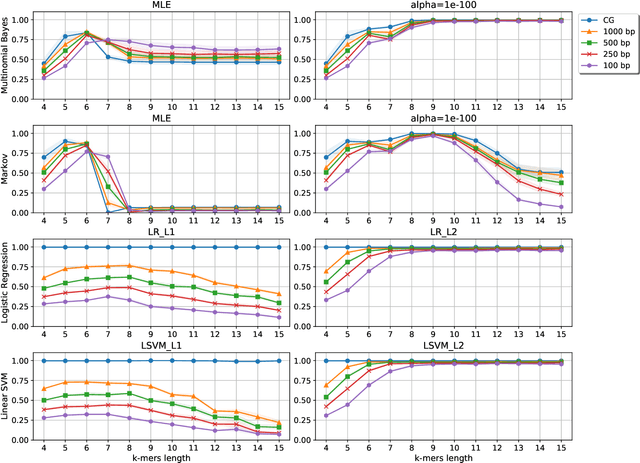

Viral sequence classification is an important task in pathogen detection, epidemiological surveys and evolutionary studies. Statistical learning methods are widely used to classify and identify viral sequences in samples from environments. These methods face several challenges associated with the nature and properties of viral genomes such as recombination, mutation rate and diversity. Also, new generations of sequencing technologies rise other difficulties by generating massive amounts of fragmented sequences. While linear classifiers are often used to classify viruses, there is a lack of exploration of the accuracy space of existing models in the context of alignment free approaches. In this study, we present an exhaustive assessment procedure exploring the power of linear classifiers in genotyping and subtyping partial and complete genomes. It is applied to the Hepatitis C viruses (HCV). Several variables are considered in this investigation such as classifier types (generative and discriminative) and their hyper-parameters (smoothing value and regularization penalty function), the classification task (genotyping and subtyping), the length of the tested sequences (partial and complete) and the length of k-mer words. Overall, several classifiers perform well given a set of precise combination of the experimental variables mentioned above. Finally, we provide the procedure and benchmark data to allow for more robust assessment of classification from virus genomes.
Toward an Efficient Multi-class Classification in an Open Universe
Mar 01, 2018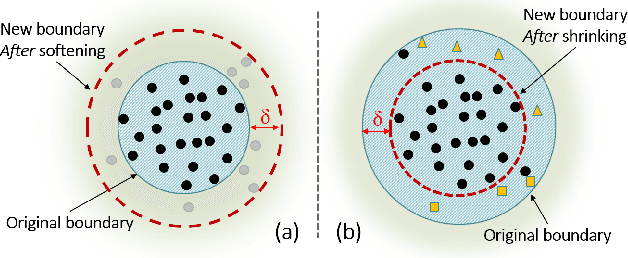
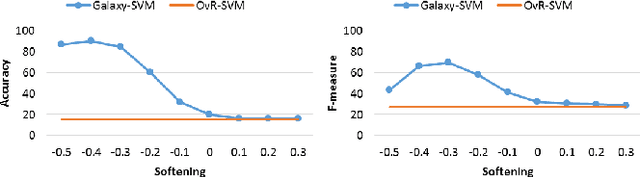

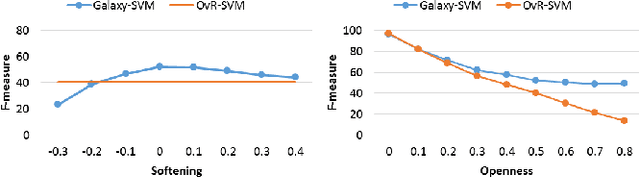
Classification is a fundamental task in machine learning and data mining. Existing classification methods are designed to classify unknown instances within a set of previously known training classes. Such a classification takes the form of a prediction within a closed-set of classes. However, a more realistic scenario that fits real-world applications is to consider the possibility of encountering instances that do not belong to any of the training classes, $i.e.$, an open-set classification. In such situation, existing closed-set classifiers will assign a training label to these instances resulting in a misclassification. In this paper, we introduce Galaxy-X, a novel multi-class classification approach for open-set recognition problems. For each class of the training set, Galaxy-X creates a minimum bounding hyper-sphere that encompasses the distribution of the class by enclosing all of its instances. In such manner, our method is able to distinguish instances resembling previously seen classes from those that are of unknown ones. To adequately evaluate open-set classification, we introduce a novel evaluation procedure. Experimental results on benchmark datasets show the efficiency of our approach in classifying novel instances from known as well as unknown classes.
ProtNN: Fast and Accurate Nearest Neighbor Protein Function Prediction based on Graph Embedding in Structural and Topological Space
Jan 25, 2016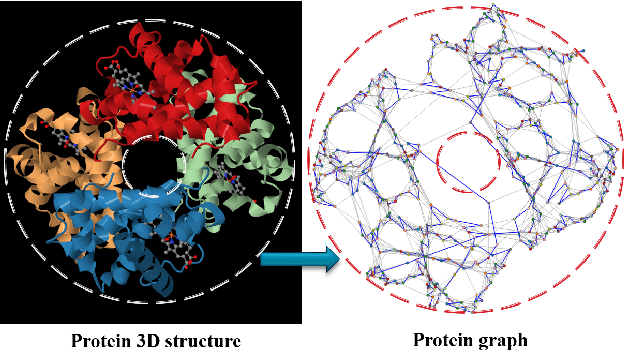
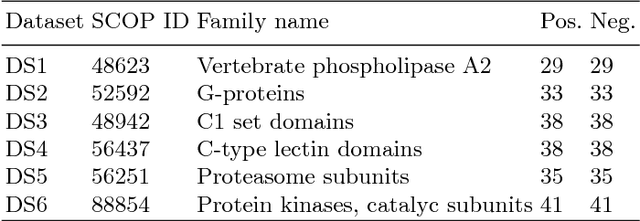
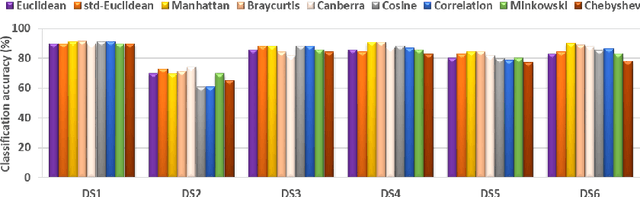

Studying the function of proteins is important for understanding the molecular mechanisms of life. The number of publicly available protein structures has increasingly become extremely large. Still, the determination of the function of a protein structure remains a difficult, costly, and time consuming task. The difficulties are often due to the essential role of spatial and topological structures in the determination of protein functions in living cells. In this paper, we propose ProtNN, a novel approach for protein function prediction. Given an unannotated protein structure and a set of annotated proteins, ProtNN finds the nearest neighbor annotated structures based on protein-graph pairwise similarities. Given a query protein, ProtNN finds the nearest neighbor reference proteins based on a graph representation model and a pairwise similarity between vector embedding of both query and reference protein-graphs in structural and topological spaces. ProtNN assigns to the query protein the function with the highest number of votes across the set of k nearest neighbor reference proteins, where k is a user-defined parameter. Experimental evaluation demonstrates that ProtNN is able to accurately classify several datasets in an extremely fast runtime compared to state-of-the-art approaches. We further show that ProtNN is able to scale up to a whole PDB dataset in a single-process mode with no parallelization, with a gain of thousands order of magnitude of runtime compared to state-of-the-art approaches.