 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrior Density Learning in Variational Bayesian Phylogenetic Parameters Inference
Feb 06, 2023The advances in variational inference are providing promising paths in Bayesian estimation problems. These advances make variational phylogenetic inference an alternative approach to Markov Chain Monte Carlo methods for approximating the phylogenetic posterior. However, one of the main drawbacks of such approaches is the modelling of the prior through fixed distributions, which could bias the posterior approximation if they are distant from the current data distribution. In this paper, we propose an approach and an implementation framework to relax the rigidity of the prior densities by learning their parameters using a gradient-based method and a neural network-based parameterization. We applied this approach for branch lengths and evolutionary parameters estimation under several Markov chain substitution models. The results of performed simulations show that the approach is powerful in estimating branch lengths and evolutionary model parameters. They also show that a flexible prior model could provide better results than a predefined prior model. Finally, the results highlight that using neural networks improves the initialization of the optimization of the prior density parameters.
EvoVGM: A Deep Variational Generative Model for Evolutionary Parameter Estimation
May 25, 2022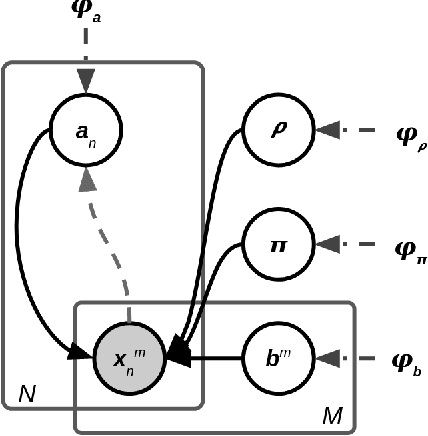
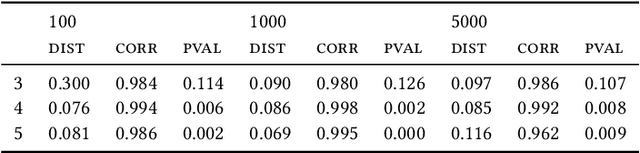
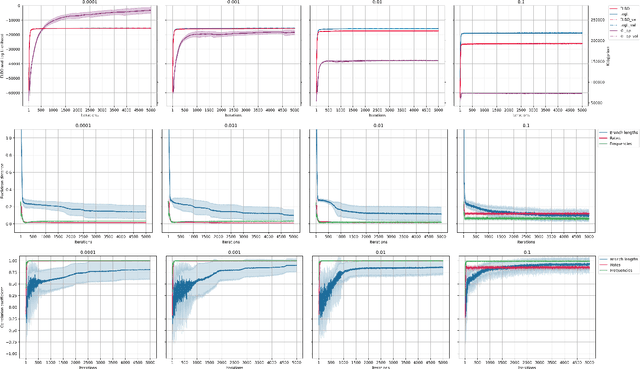
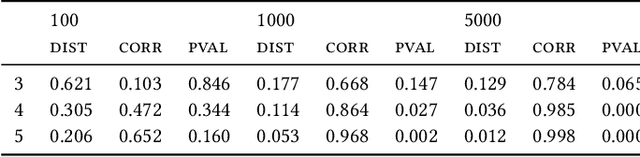
Most evolutionary-oriented deep generative models do not explicitly consider the underlying evolutionary dynamics of biological sequences as it is performed within the Bayesian phylogenetic inference framework. In this study, we propose a method for a deep variational Bayesian generative model that jointly approximates the true posterior of local biological evolutionary parameters and generates sequence alignments. Moreover, it is instantiated and tuned for continuous-time Markov chain substitution models such as JC69 and GTR. We train the model via a low-variance variational objective function and a gradient ascent algorithm. Here, we show the consistency and effectiveness of the method on synthetic sequence alignments simulated with several evolutionary scenarios and on a real virus sequence alignment.
Statistical Linear Models in Virus Genomic Alignment-free Classification: Application to Hepatitis C Viruses
Nov 06, 2019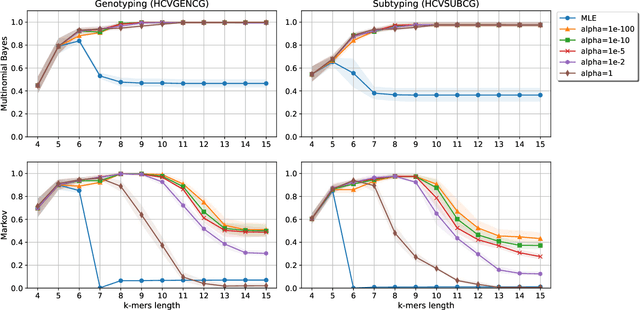
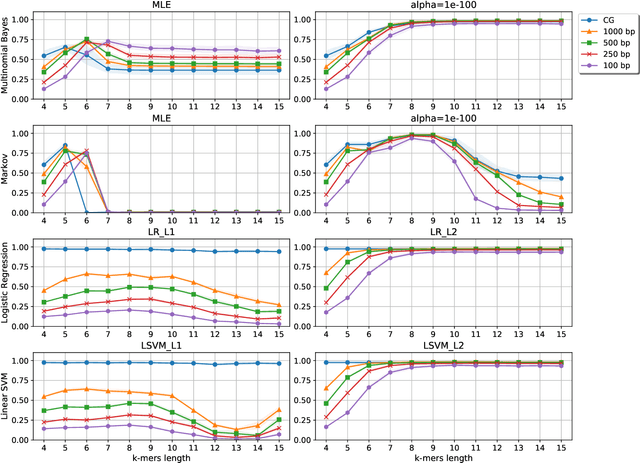
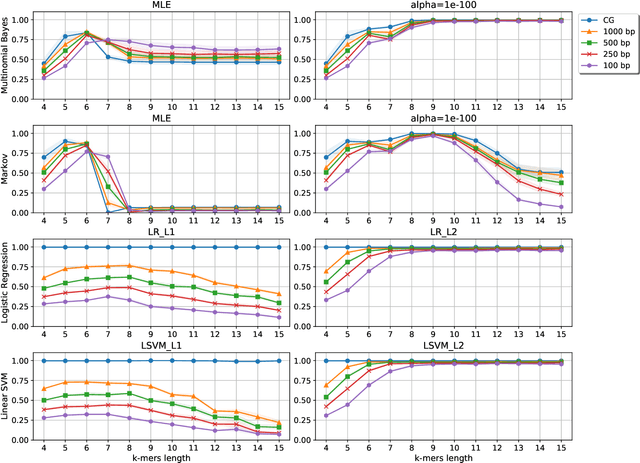

Viral sequence classification is an important task in pathogen detection, epidemiological surveys and evolutionary studies. Statistical learning methods are widely used to classify and identify viral sequences in samples from environments. These methods face several challenges associated with the nature and properties of viral genomes such as recombination, mutation rate and diversity. Also, new generations of sequencing technologies rise other difficulties by generating massive amounts of fragmented sequences. While linear classifiers are often used to classify viruses, there is a lack of exploration of the accuracy space of existing models in the context of alignment free approaches. In this study, we present an exhaustive assessment procedure exploring the power of linear classifiers in genotyping and subtyping partial and complete genomes. It is applied to the Hepatitis C viruses (HCV). Several variables are considered in this investigation such as classifier types (generative and discriminative) and their hyper-parameters (smoothing value and regularization penalty function), the classification task (genotyping and subtyping), the length of the tested sequences (partial and complete) and the length of k-mer words. Overall, several classifiers perform well given a set of precise combination of the experimental variables mentioned above. Finally, we provide the procedure and benchmark data to allow for more robust assessment of classification from virus genomes.