 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA multiple instance learning approach for sequence data with across bag dependencies
Jan 30, 2016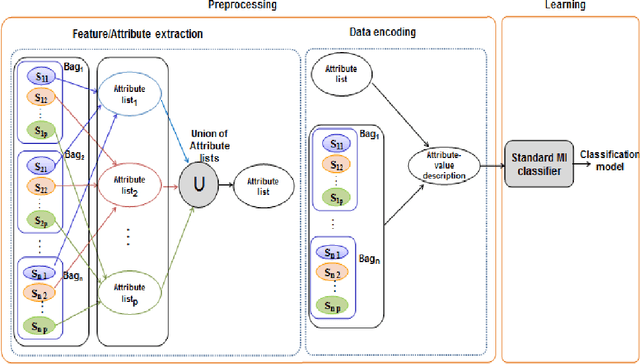
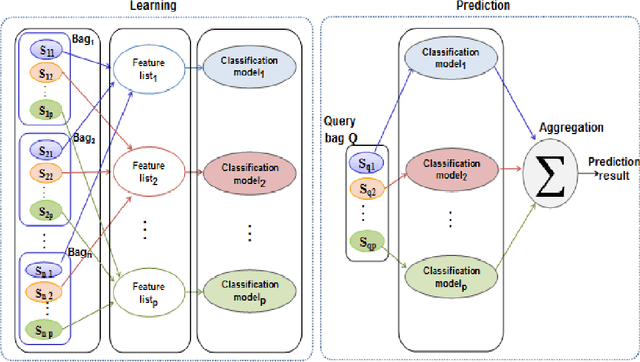
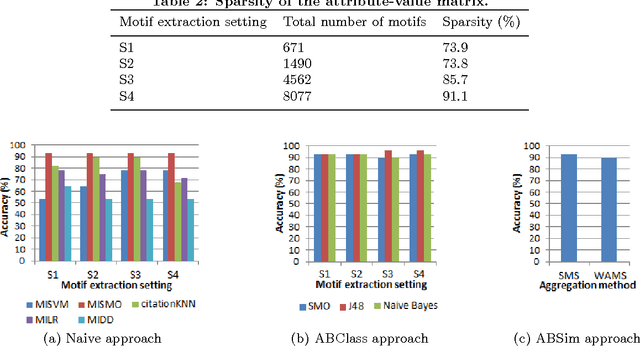
In Multiple Instance Learning (MIL) problem for sequence data, the learning data consist of a set of bags where each bag contains a set of instances/sequences. In many real world applications such as bioinformatics, web mining, and text mining, comparing a random couple of sequences makes no sense. In fact, each instance of each bag may have structural and/or temporal relation with other instances in other bags. Thus, the classification task should take into account the relation between semantically related instances across bags. In this paper, we present two novel MIL approaches for sequence data classification: (1) ABClass and (2) ABSim. In ABClass, each sequence is represented by one vector of attributes. For each sequence of the unknown bag, a discriminative classifier is applied in order to compute a partial classification result. Then, an aggregation method is applied to these partial results in order to generate the final result. In ABSim, we use a similarity measure between each sequence of the unknown bag and the corresponding sequences in the learning bags. An unknown bag is labeled with the bag that presents more similar sequences. We applied both approaches to the problem of bacterial Ionizing Radiation Resistance (IRR) prediction. We evaluated and discussed the proposed approaches on well known Ionizing Radiation Resistance Bacteria (IRRB) and Ionizing Radiation Sensitive Bacteria (IRSB) represented by primary structure of basal DNA repair proteins. The experimental results show that both ABClass and ABSim approaches are efficient.
Feature extraction in protein sequences classification : a new stability measure
Dec 05, 2012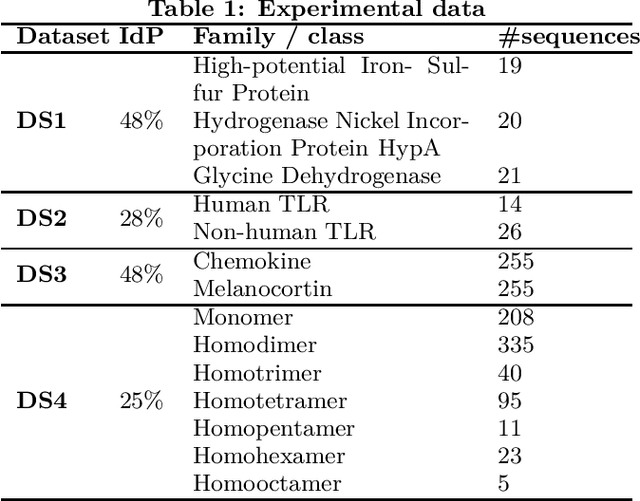
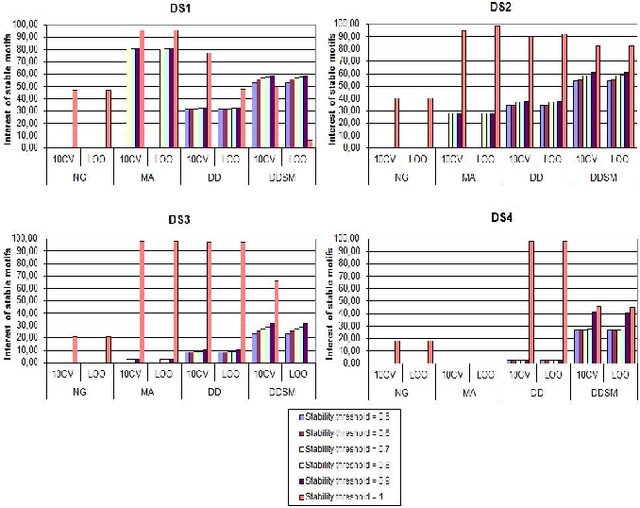
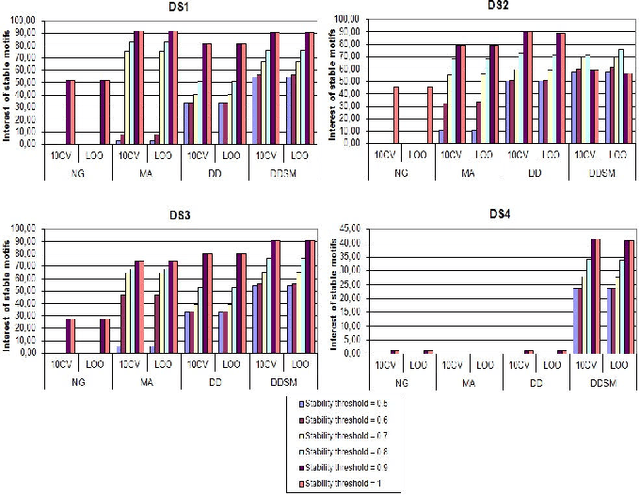
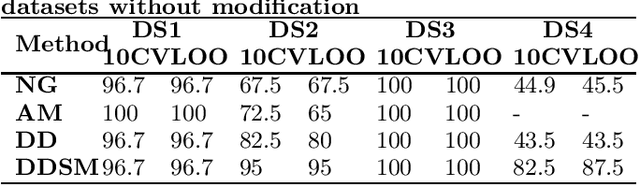
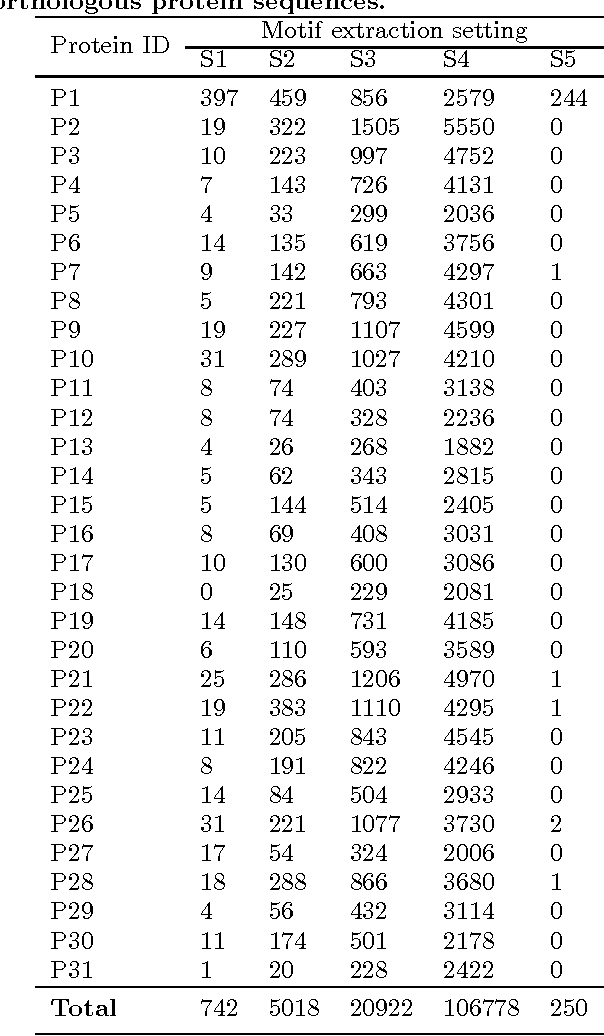
Feature extraction is an unavoidable task, especially in the critical step of preprocessing biological sequences. This step consists for example in transforming the biological sequences into vectors of motifs where each motif is a subsequence that can be seen as a property (or attribute) characterizing the sequence. Hence, we obtain an object-property table where objects are sequences and properties are motifs extracted from sequences. This output can be used to apply standard machine learning tools to perform data mining tasks such as classification. Several previous works have described feature extraction methods for bio-sequence classification, but none of them discussed the robustness of these methods when perturbing the input data. In this work, we introduce the notion of stability of the generated motifs in order to study the robustness of motif extraction methods. We express this robustness in terms of the ability of the method to reveal any change occurring in the input data and also its ability to target the interesting motifs. We use these criteria to evaluate and experimentally compare four existing extraction methods for biological sequences.