 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Combinatorial Generalization for Catalysts: A Kohn-Sham Charge-Density Approach
Oct 28, 2023



The Kohn-Sham equations underlie many important applications such as the discovery of new catalysts. Recent machine learning work on catalyst modeling has focused on prediction of the energy, but has so far not yet demonstrated significant out-of-distribution generalization. Here we investigate another approach based on the pointwise learning of the Kohn-Sham charge-density. On a new dataset of bulk catalysts with charge densities, we show density models can generalize to new structures with combinations of elements not seen at train time, a form of combinatorial generalization. We show that over 80% of binary and ternary test cases achieve faster convergence than standard baselines in Density Functional Theory, amounting to an average reduction of 13% in the number of iterations required to reach convergence, which may be of independent interest. Our results suggest that density learning is a viable alternative, trading greater inference costs for a step towards combinatorial generalization, a key property for applications.
A Comprehensive Study of Image Classification Model Sensitivity to Foregrounds, Backgrounds, and Visual Attributes
Jan 26, 2022
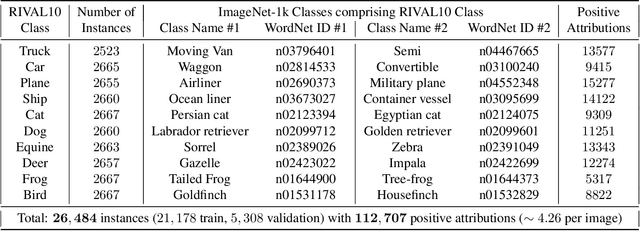
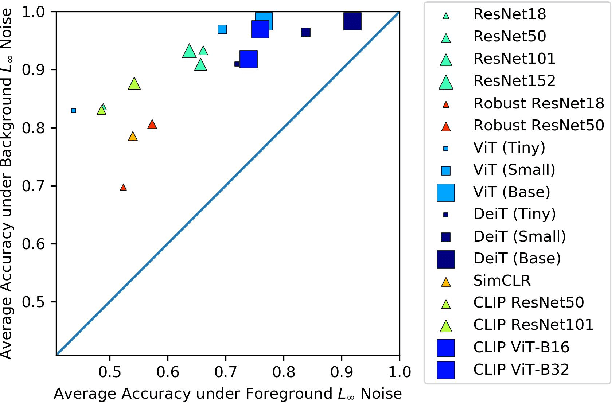

While datasets with single-label supervision have propelled rapid advances in image classification, additional annotations are necessary in order to quantitatively assess how models make predictions. To this end, for a subset of ImageNet samples, we collect segmentation masks for the entire object and $18$ informative attributes. We call this dataset RIVAL10 (RIch Visual Attributes with Localization), consisting of roughly $26k$ instances over $10$ classes. Using RIVAL10, we evaluate the sensitivity of a broad set of models to noise corruptions in foregrounds, backgrounds and attributes. In our analysis, we consider diverse state-of-the-art architectures (ResNets, Transformers) and training procedures (CLIP, SimCLR, DeiT, Adversarial Training). We find that, somewhat surprisingly, in ResNets, adversarial training makes models more sensitive to the background compared to foreground than standard training. Similarly, contrastively-trained models also have lower relative foreground sensitivity in both transformers and ResNets. Lastly, we observe intriguing adaptive abilities of transformers to increase relative foreground sensitivity as corruption level increases. Using saliency methods, we automatically discover spurious features that drive the background sensitivity of models and assess alignment of saliency maps with foregrounds. Finally, we quantitatively study the attribution problem for neural features by comparing feature saliency with ground-truth localization of semantic attributes.
The Intrinsic Dimension of Images and Its Impact on Learning
Apr 18, 2021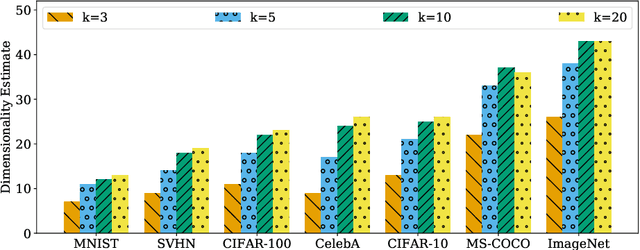



It is widely believed that natural image data exhibits low-dimensional structure despite the high dimensionality of conventional pixel representations. This idea underlies a common intuition for the remarkable success of deep learning in computer vision. In this work, we apply dimension estimation tools to popular datasets and investigate the role of low-dimensional structure in deep learning. We find that common natural image datasets indeed have very low intrinsic dimension relative to the high number of pixels in the images. Additionally, we find that low dimensional datasets are easier for neural networks to learn, and models solving these tasks generalize better from training to test data. Along the way, we develop a technique for validating our dimension estimation tools on synthetic data generated by GANs allowing us to actively manipulate the intrinsic dimension by controlling the image generation process. Code for our experiments may be found here https://github.com/ppope/dimensions.
Adversarial Robustness of Flow-Based Generative Models
Nov 20, 2019
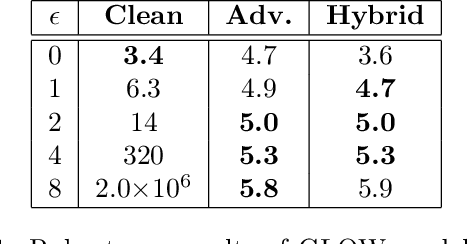
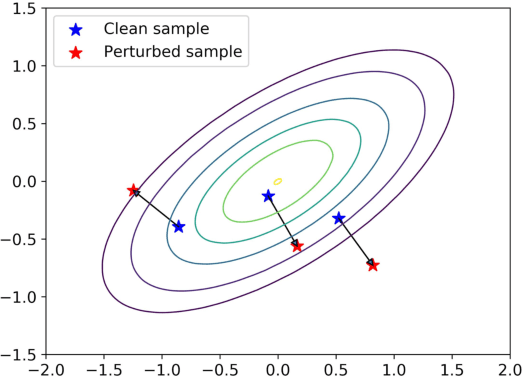
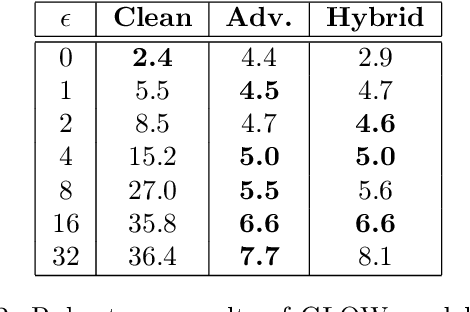
Flow-based generative models leverage invertible generator functions to fit a distribution to the training data using maximum likelihood. Despite their use in several application domains, robustness of these models to adversarial attacks has hardly been explored. In this paper, we study adversarial robustness of flow-based generative models both theoretically (for some simple models) and empirically (for more complex ones). First, we consider a linear flow-based generative model and compute optimal sample-specific and universal adversarial perturbations that maximally decrease the likelihood scores. Using this result, we study the robustness of the well-known adversarial training procedure, where we characterize the fundamental trade-off between model robustness and accuracy. Next, we empirically study the robustness of two prominent deep, non-linear, flow-based generative models, namely GLOW and RealNVP. We design two types of adversarial attacks; one that minimizes the likelihood scores of in-distribution samples, while the other that maximizes the likelihood scores of out-of-distribution ones. We find that GLOW and RealNVP are extremely sensitive to both types of attacks. Finally, using a hybrid adversarial training procedure, we significantly boost the robustness of these generative models.
Discovering Molecular Functional Groups Using Graph Convolutional Neural Networks
Dec 06, 2018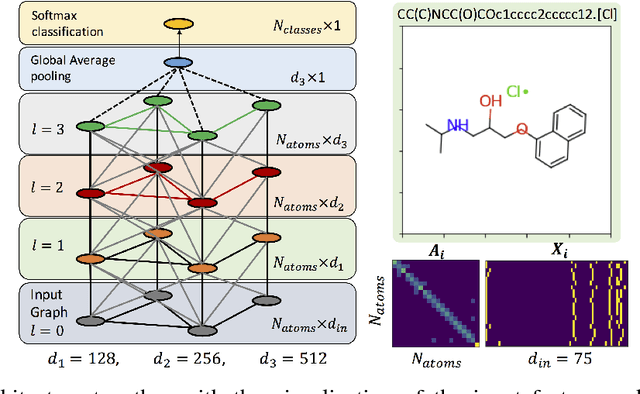
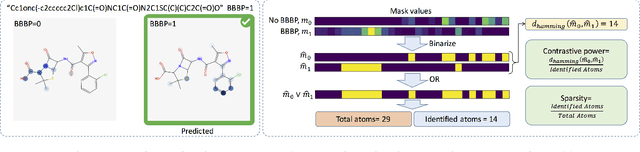
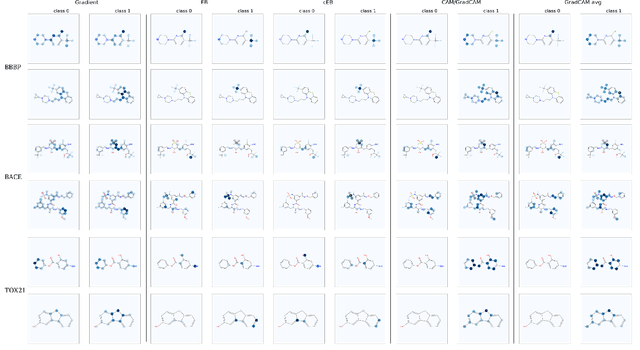
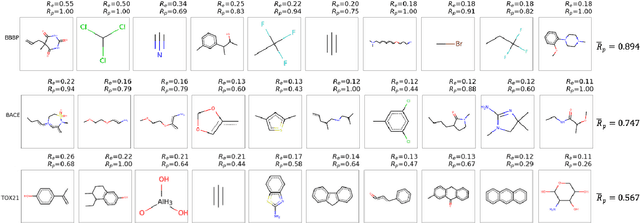
Functional groups (FGs) serve as a foundation for analyzing chemical properties of organic molecules. Automatic discovery of FGs will impact various fields of research, including medicinal chemistry, by reducing the amount of lab experiments required for discovery or synthesis of new molecules. Here, we investigate methods based on graph convolutional neural networks (GCNNs) for localizing FGs that contribute to specific chemical properties. Molecules are modeled as undirected graphs with atoms as nodes and bonds as edges. Using this graph structure, we trained GCNNs in a supervised way on experimentally-validated molecular training sets to predict specific chemical properties, e.g., toxicity. Upon learning a GCNN, we analyzed its activation patterns to automatically identify FGs using four different methods: gradient-based saliency maps, Class Activation Mapping (CAM), gradient-weighted CAM (Grad-CAM), and Excitation Back-Propagation. We evaluated the contrastive power of these methods with respect to the specificity of the identified molecular substructures and their relevance for chemical functions. Grad- CAM had the highest contrastive power and generated qualitatively the best FGs. This work paves the way for automatic analysis and design of new molecules.