 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDiscovering Molecular Functional Groups Using Graph Convolutional Neural Networks
Paper and Code
Dec 06, 2018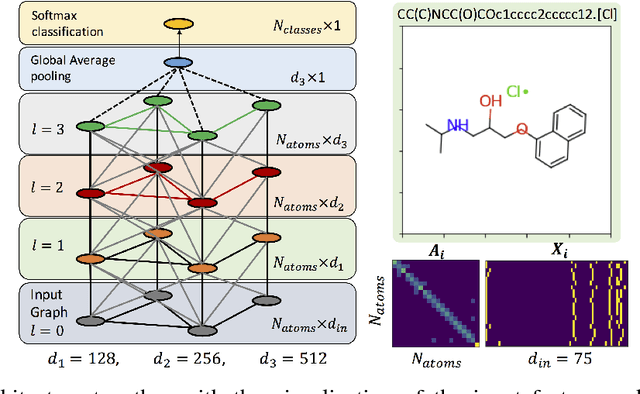
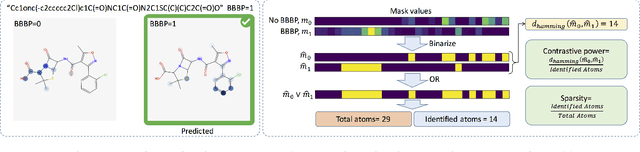
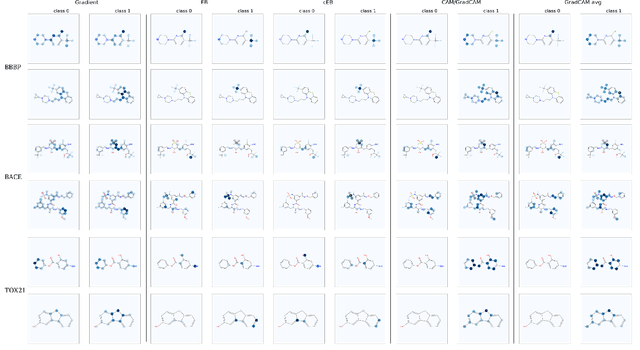
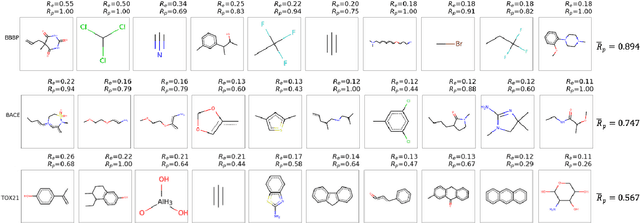
Functional groups (FGs) serve as a foundation for analyzing chemical properties of organic molecules. Automatic discovery of FGs will impact various fields of research, including medicinal chemistry, by reducing the amount of lab experiments required for discovery or synthesis of new molecules. Here, we investigate methods based on graph convolutional neural networks (GCNNs) for localizing FGs that contribute to specific chemical properties. Molecules are modeled as undirected graphs with atoms as nodes and bonds as edges. Using this graph structure, we trained GCNNs in a supervised way on experimentally-validated molecular training sets to predict specific chemical properties, e.g., toxicity. Upon learning a GCNN, we analyzed its activation patterns to automatically identify FGs using four different methods: gradient-based saliency maps, Class Activation Mapping (CAM), gradient-weighted CAM (Grad-CAM), and Excitation Back-Propagation. We evaluated the contrastive power of these methods with respect to the specificity of the identified molecular substructures and their relevance for chemical functions. Grad- CAM had the highest contrastive power and generated qualitatively the best FGs. This work paves the way for automatic analysis and design of new molecules.