 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeD2C: Unlocking the Potential of Continuous Autoregressive Image Generation with Discrete Tokens
Mar 21, 2025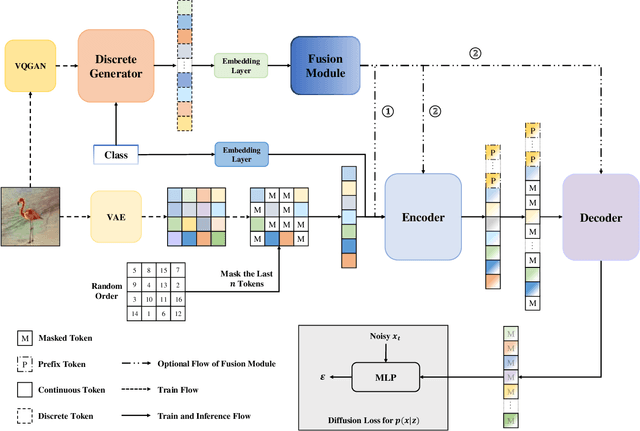
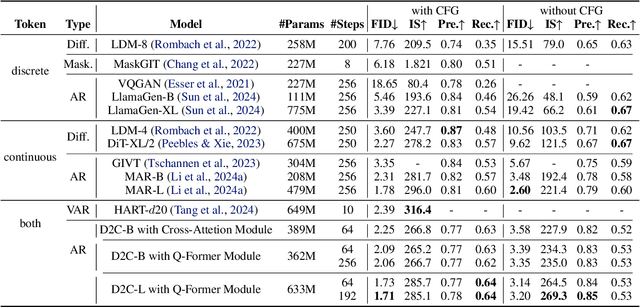
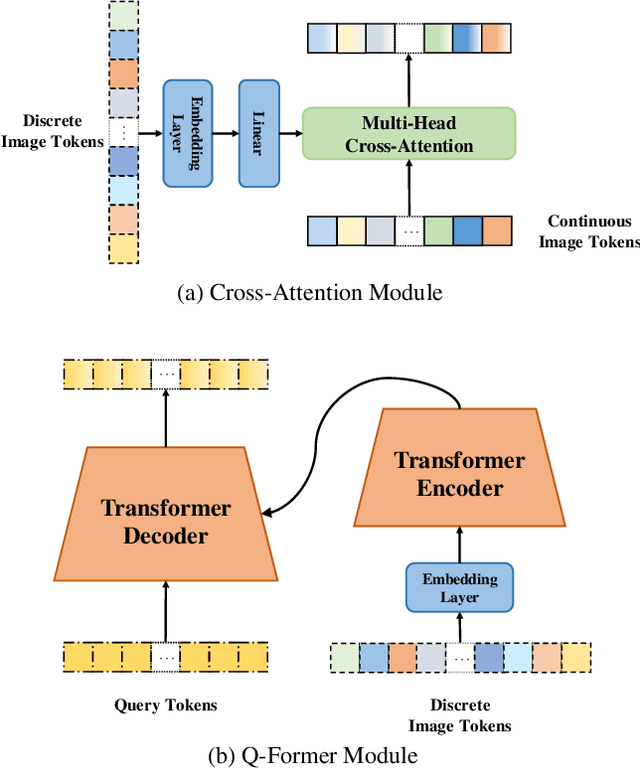
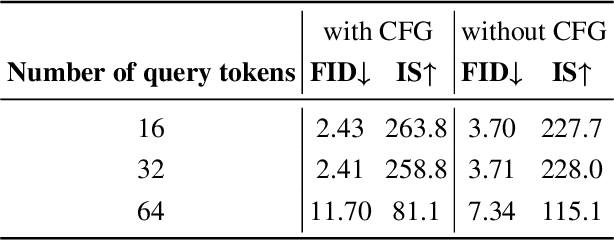
In the domain of image generation, latent-based generative models occupy a dominant status; however, these models rely heavily on image tokenizer. To meet modeling requirements, autoregressive models possessing the characteristics of scalability and flexibility embrace a discrete-valued tokenizer, but face the challenge of poor image generation quality. In contrast, diffusion models take advantage of the continuous-valued tokenizer to achieve better generation quality but are subject to low efficiency and complexity. The existing hybrid models are mainly to compensate for information loss and simplify the diffusion learning process. The potential of merging discrete-valued and continuous-valued tokens in the field of image generation has not yet been explored. In this paper, we propose D2C, a novel two-stage method to enhance model generation capacity. In the first stage, the discrete-valued tokens representing coarse-grained image features are sampled by employing a small discrete-valued generator. Then in the second stage, the continuous-valued tokens representing fine-grained image features are learned conditioned on the discrete token sequence. In addition, we design two kinds of fusion modules for seamless interaction. On the ImageNet-256 benchmark, extensive experiment results validate that our model achieves superior performance compared with several continuous-valued and discrete-valued generative models on the class-conditional image generation tasks.
A New Statistical Framework for Genetic Pleiotropic Analysis of High Dimensional Phenotype Data
Dec 03, 2015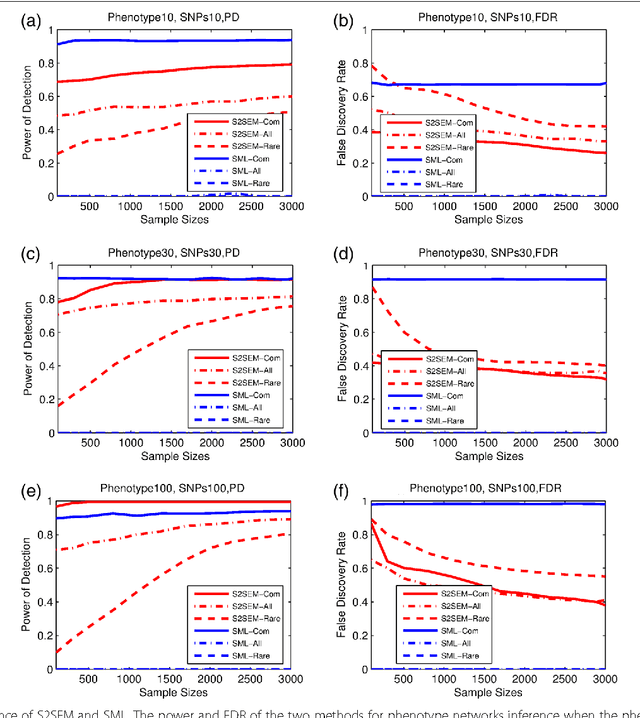
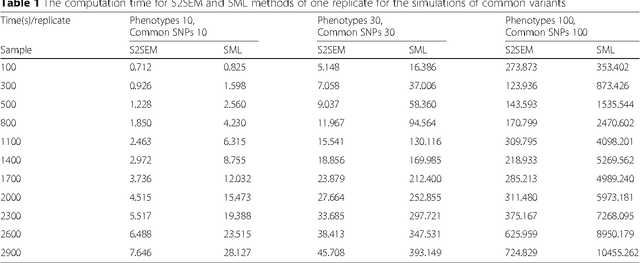
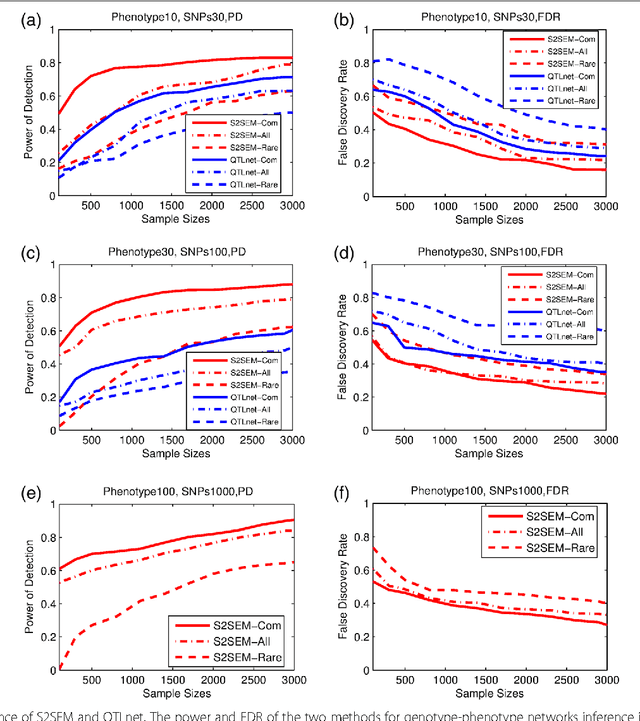
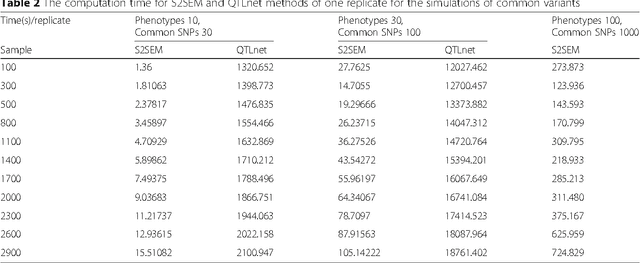
The widely used genetic pleiotropic analysis of multiple phenotypes are often designed for examining the relationship between common variants and a few phenotypes. They are not suited for both high dimensional phenotypes and high dimensional genotype (next-generation sequencing) data. To overcome these limitations, we develop sparse structural equation models (SEMs) as a general framework for a new paradigm of genetic analysis of multiple phenotypes. To incorporate both common and rare variants into the analysis, we extend the traditional multivariate SEMs to sparse functional SEMs. To deal with high dimensional phenotype and genotype data, we employ functional data analysis and the alternative direction methods of multiplier (ADMM) techniques to reduce data dimension and improve computational efficiency. Using large scale simulations we showed that the proposed methods have higher power to detect true causal genetic pleiotropic structure than other existing methods. Simulations also demonstrate that the gene-based pleiotropic analysis has higher power than the single variant-based pleiotropic analysis. The proposed method is applied to exome sequence data from the NHLBI Exome Sequencing Project (ESP) with 11 phenotypes, which identifies a network with 137 genes connected to 11 phenotypes and 341 edges. Among them, 114 genes showed pleiotropic genetic effects and 45 genes were reported to be associated with phenotypes in the analysis or other cardiovascular disease (CVD) related phenotypes in the literature.