 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIterative Rule Extension for Logic Analysis of Data: an MILP-based heuristic to derive interpretable binary classification from large datasets
Oct 25, 2021
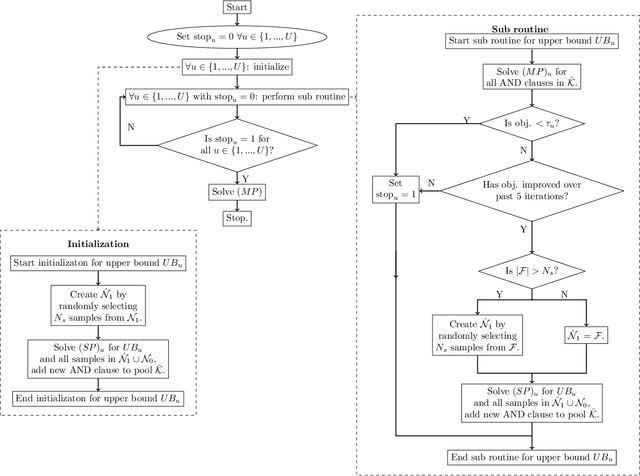
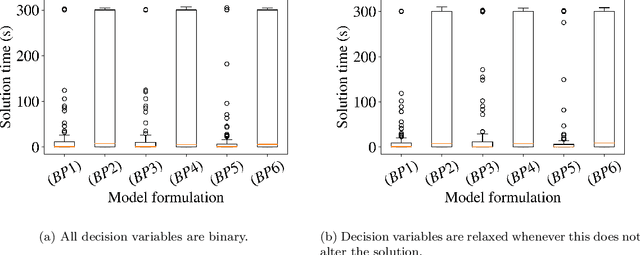
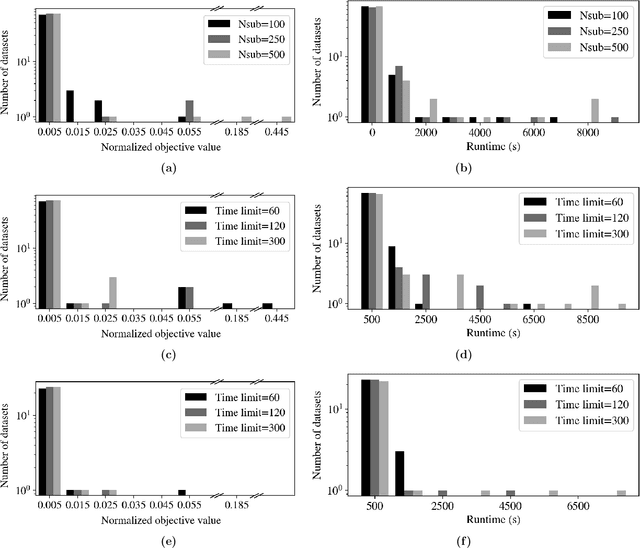
Data-driven decision making is rapidly gaining popularity, fueled by the ever-increasing amounts of available data and encouraged by the development of models that can identify beyond linear input-output relationships. Simultaneously the need for interpretable prediction- and classification methods is increasing, as this improves both our trust in these models and the amount of information we can abstract from data. An important aspect of this interpretability is to obtain insight in the sensitivity-specificity trade-off constituted by multiple plausible input-output relationships. These are often shown in a receiver operating characteristic (ROC) curve. These developments combined lead to the need for a method that can abstract complex yet interpretable input-output relationships from large data, i.e. data containing large numbers of samples and sample features. Boolean phrases in disjunctive normal form (DNF) are highly suitable for explaining non-linear input-output relationships in a comprehensible way. Mixed integer linear programming (MILP) can be used to abstract these Boolean phrases from binary data, though its computational complexity prohibits the analysis of large datasets. This work presents IRELAND, an algorithm that allows for abstracting Boolean phrases in DNF from data with up to 10,000 samples and sample characteristics. The results show that for large datasets IRELAND outperforms the current state-of-the-art and can find solutions for datasets where current models run out of memory or need excessive runtimes. Additionally, by construction IRELAND allows for an efficient computation of the sensitivity-specificity trade-off curve, allowing for further understanding of the underlying input-output relationship.
A Drug Recommendation System for cancer cell lines
Dec 24, 2019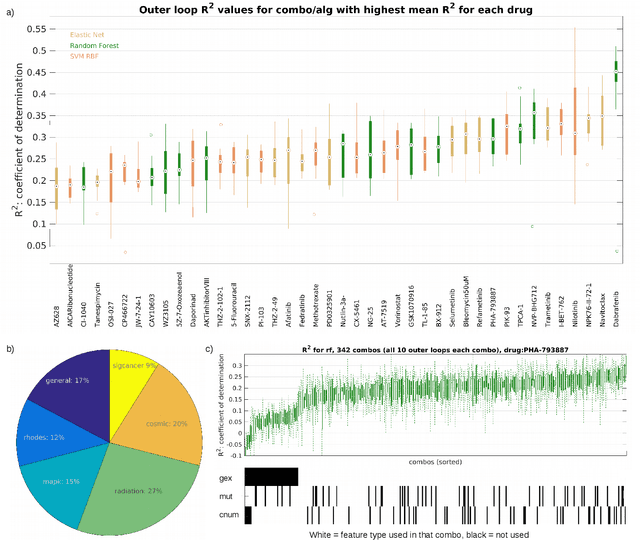
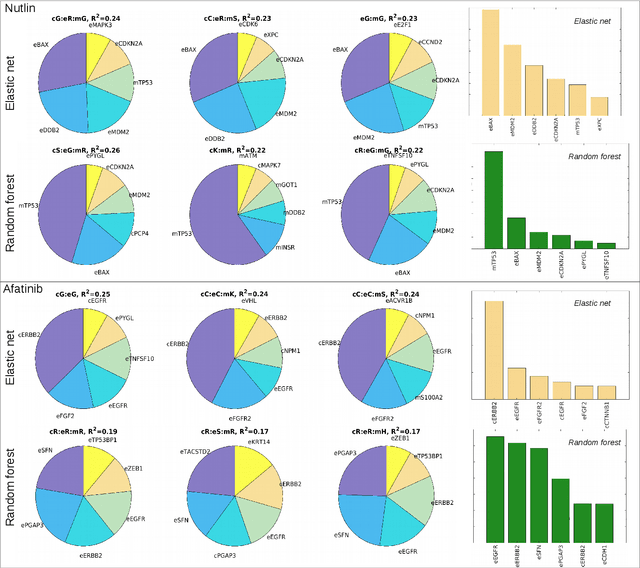
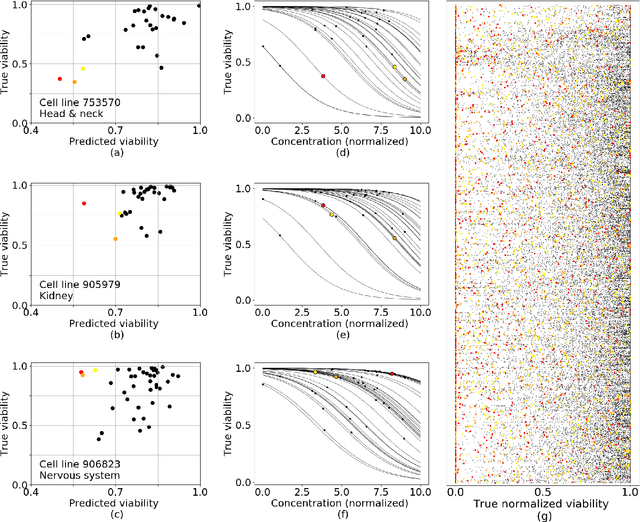
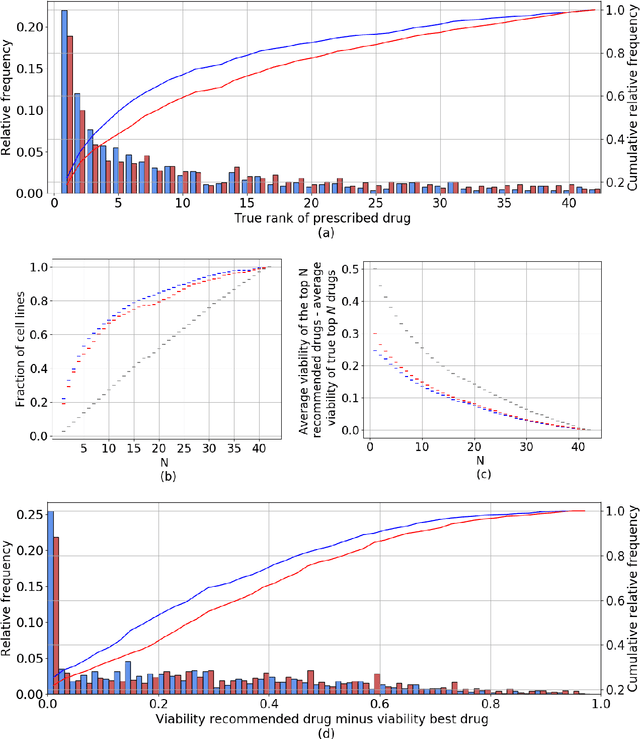
Personalizing drug prescriptions in cancer care based on genomic information requires associating genomic markers with treatment effects. This is an unsolved challenge requiring genomic patient data in yet unavailable volumes as well as appropriate quantitative methods. We attempt to solve this challenge for an experimental proxy for which sufficient data is available: 42 drugs tested on 1018 cancer cell lines. Our goal is to develop a method to identify the drug that is most promising based on a cell line's genomic information. For this, we need to identify for each drug the machine learning method, choice of hyperparameters and genomic features for optimal predictive performance. We extensively compare combinations of gene sets (both curated and random), genetic features, and machine learning algorithms for all 42 drugs. For each drug, the best performing combination (considering only the curated gene sets) is selected. We use these top model parameters for each drug to build and demonstrate a Drug Recommendation System (Dr.S). Insights resulting from this analysis are formulated as best practices for developing drug recommendation systems. The complete software system, called the Cell Line Analyzer, is written in Python and available on github.
An image representation based convolutional network for DNA classification
Jun 13, 2018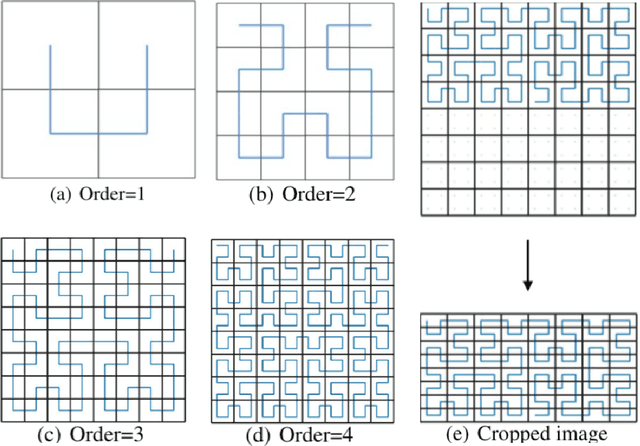
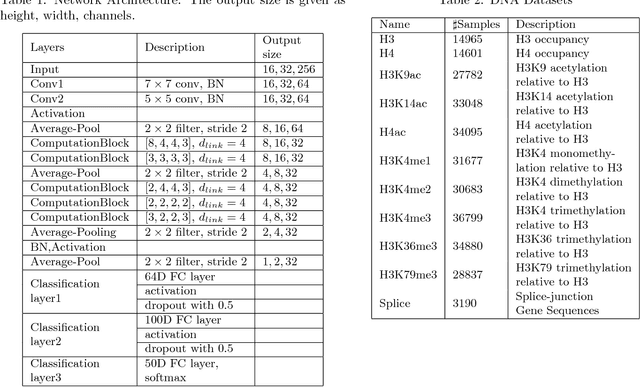
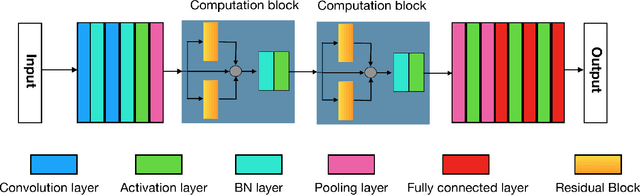
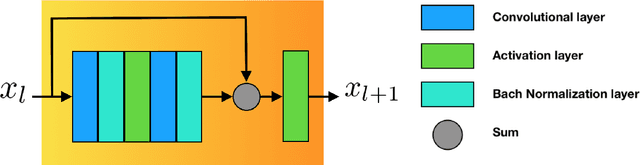
The folding structure of the DNA molecule combined with helper molecules, also referred to as the chromatin, is highly relevant for the functional properties of DNA. The chromatin structure is largely determined by the underlying primary DNA sequence, though the interaction is not yet fully understood. In this paper we develop a convolutional neural network that takes an image-representation of primary DNA sequence as its input, and predicts key determinants of chromatin structure. The method is developed such that it is capable of detecting interactions between distal elements in the DNA sequence, which are known to be highly relevant. Our experiments show that the method outperforms several existing methods both in terms of prediction accuracy and training time.