 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Drug Recommendation System for cancer cell lines
Dec 24, 2019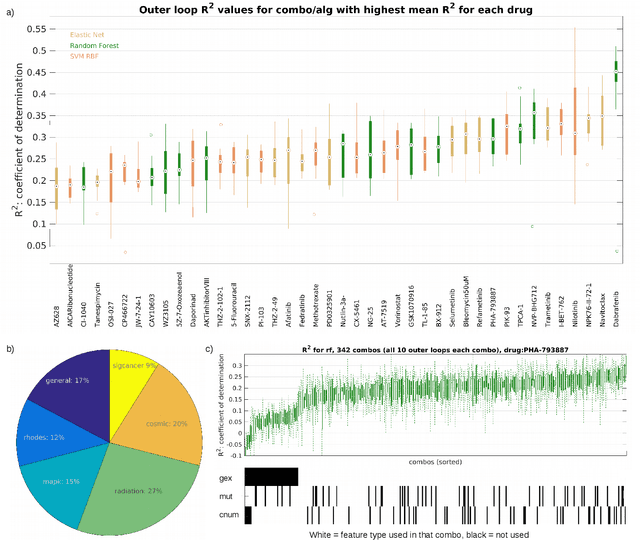
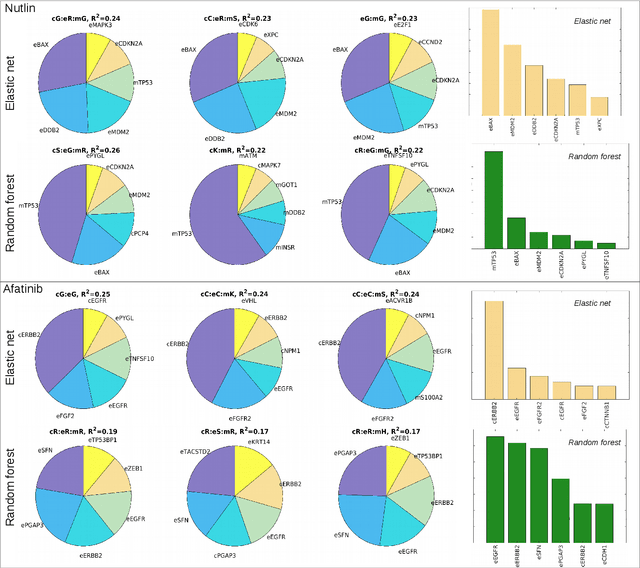
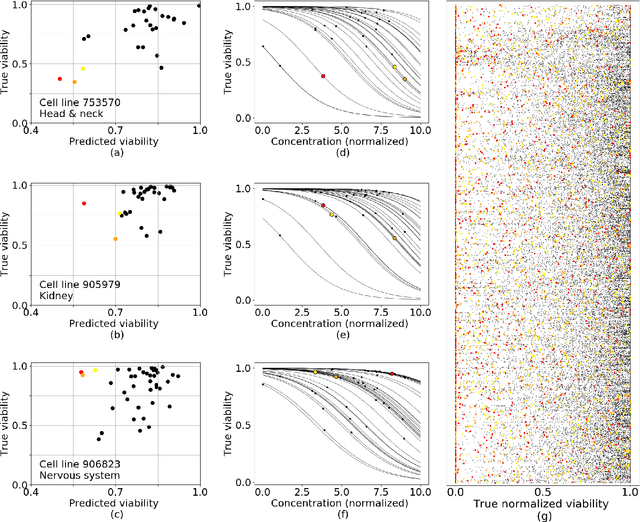
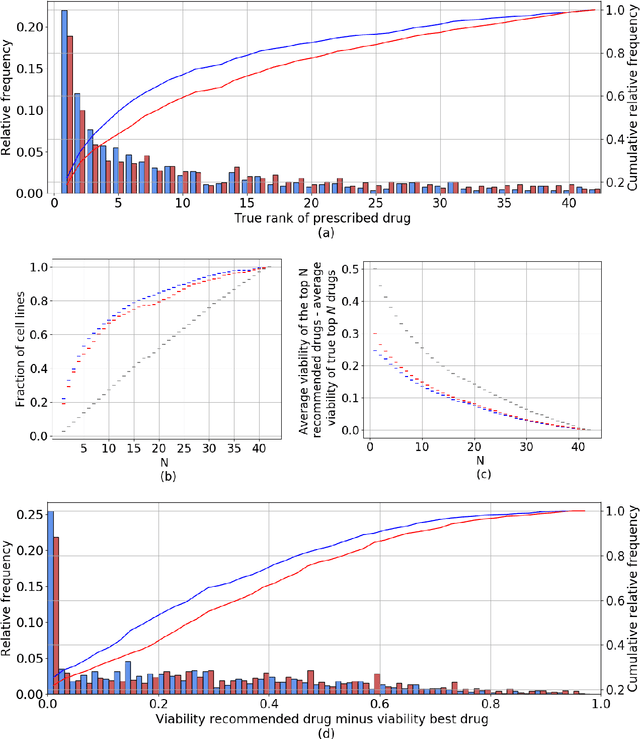
Personalizing drug prescriptions in cancer care based on genomic information requires associating genomic markers with treatment effects. This is an unsolved challenge requiring genomic patient data in yet unavailable volumes as well as appropriate quantitative methods. We attempt to solve this challenge for an experimental proxy for which sufficient data is available: 42 drugs tested on 1018 cancer cell lines. Our goal is to develop a method to identify the drug that is most promising based on a cell line's genomic information. For this, we need to identify for each drug the machine learning method, choice of hyperparameters and genomic features for optimal predictive performance. We extensively compare combinations of gene sets (both curated and random), genetic features, and machine learning algorithms for all 42 drugs. For each drug, the best performing combination (considering only the curated gene sets) is selected. We use these top model parameters for each drug to build and demonstrate a Drug Recommendation System (Dr.S). Insights resulting from this analysis are formulated as best practices for developing drug recommendation systems. The complete software system, called the Cell Line Analyzer, is written in Python and available on github.
Simulation assisted machine learning
Feb 20, 2018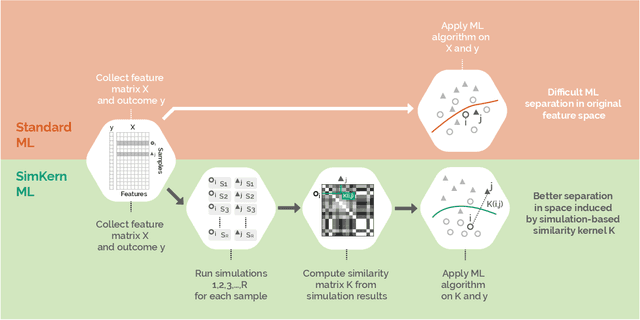
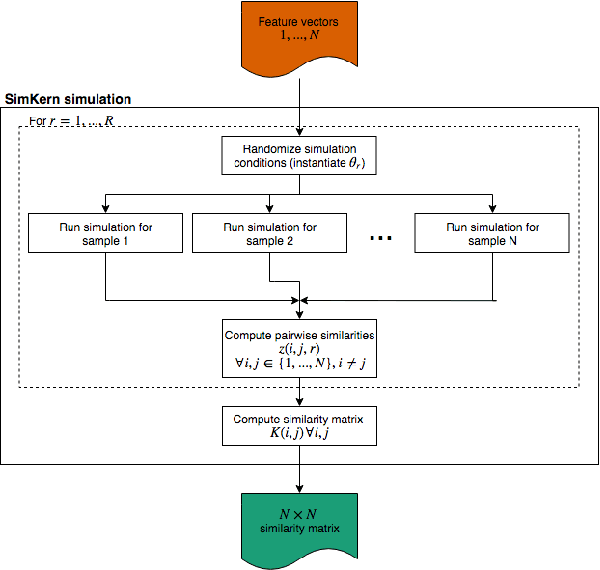
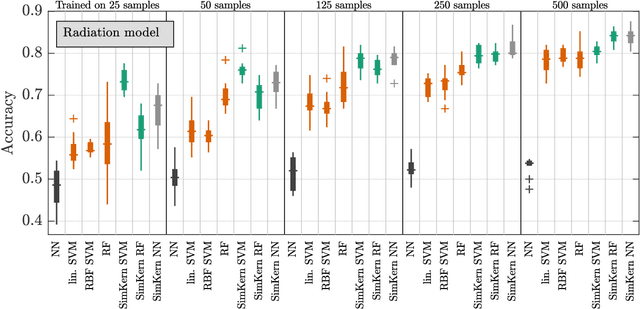
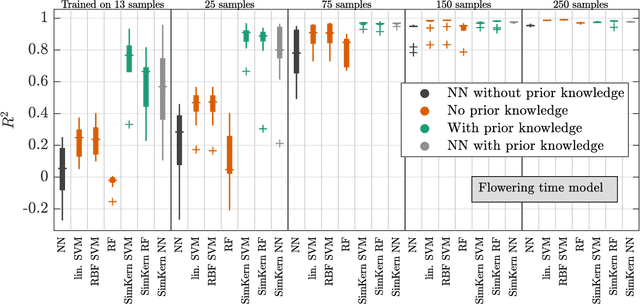
Predicting how a proposed cancer treatment will affect a given tumor can be cast as a machine learning problem, but the complexity of biological systems, the number of potentially relevant genomic and clinical features, and the lack of very large scale patient data repositories make this a unique challenge. "Pure data" approaches to this problem are underpowered to detect combinatorially complex interactions and are bound to uncover false correlations despite statistical precautions taken (1). To investigate this setting, we propose a method to integrate simulations, a strong form of prior knowledge, into machine learning, a combination which to date has been largely unexplored. The results of multiple simulations (under various uncertainty scenarios) are used to compute similarity measures between every pair of samples: sample pairs are given a high similarity score if they behave similarly under a wide range of simulation parameters. These similarity values, rather than the original high dimensional feature data, are used to train kernelized machine learning algorithms such as support vector machines, thus handling the curse-of-dimensionality that typically affects genomic machine learning. Using four synthetic datasets of complex systems--three biological models and one network flow optimization model--we demonstrate that when the number of training samples is small compared to the number of features, the simulation kernel approach dominates over no-prior-knowledge methods. In addition to biology and medicine, this approach should be applicable to other disciplines, such as weather forecasting, financial markets, and agricultural management, where predictive models are sought and informative yet approximate simulations are available. The Python SimKern software, the models (in MATLAB, Octave, and R), and the datasets are made freely available at https://github.com/davidcraft/SimKern .