 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeModel Bias in NLP -- Application to Hate Speech Classification using transfer learning techniques
Oct 11, 2021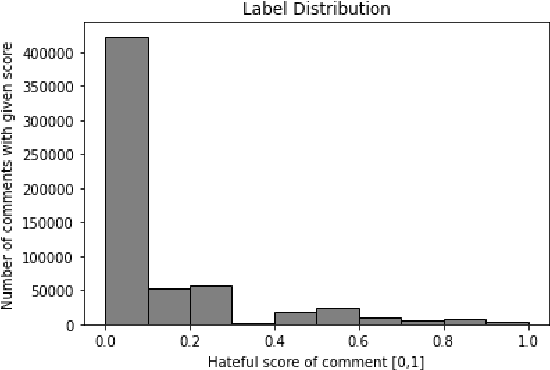
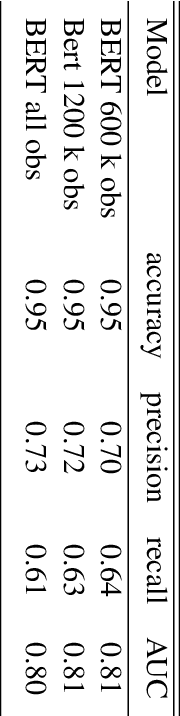
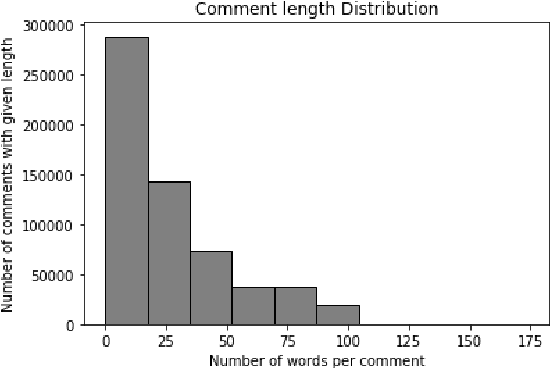
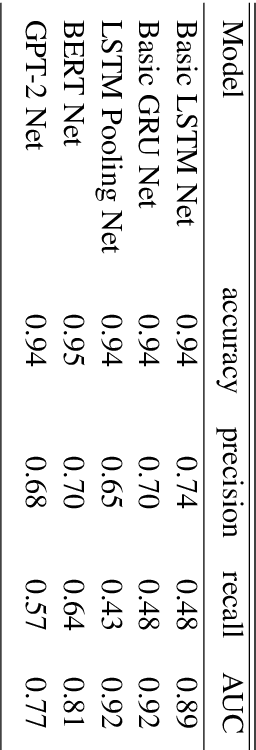
In this paper, a BERT based neural network model is applied to the JIGSAW data set in order to create a model identifying hateful and toxic comments (strictly seperated from offensive language) in online social platforms (English language), in this case Twitter. Three other neural network architectures and a GPT-2 model are also applied on the provided data set in order to compare these different models. The trained BERT model is then applied on two different data sets to evaluate its generalisation power, namely on another Twitter data set and the data set HASOC 2019 which includes Twitter and also Facebook comments; we focus on the English HASOC 2019 data. In addition, it can be shown that by fine-tuning the trained BERT model on these two data sets by applying different transfer learning scenarios via retraining partial or all layers the predictive scores improve compared to simply applying the model pre-trained on the JIGSAW data set. With our results, we get precisions from 64% to around 90% while still achieving acceptable recall values of at least lower 60s%, proving that BERT is suitable for real use cases in social platforms.
A Drug Recommendation System for cancer cell lines
Dec 24, 2019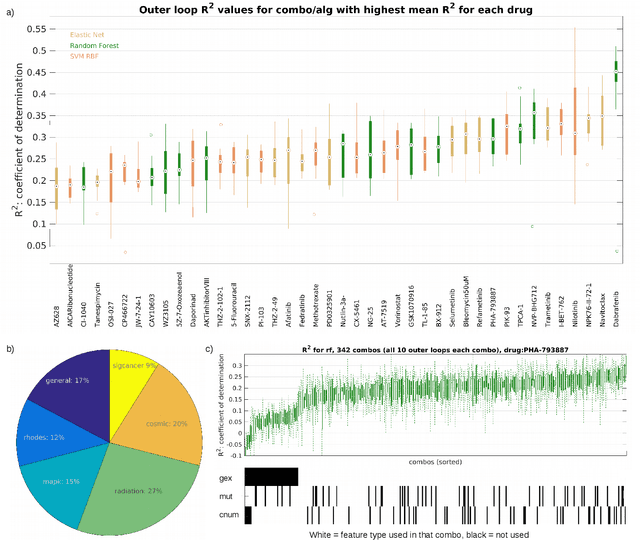
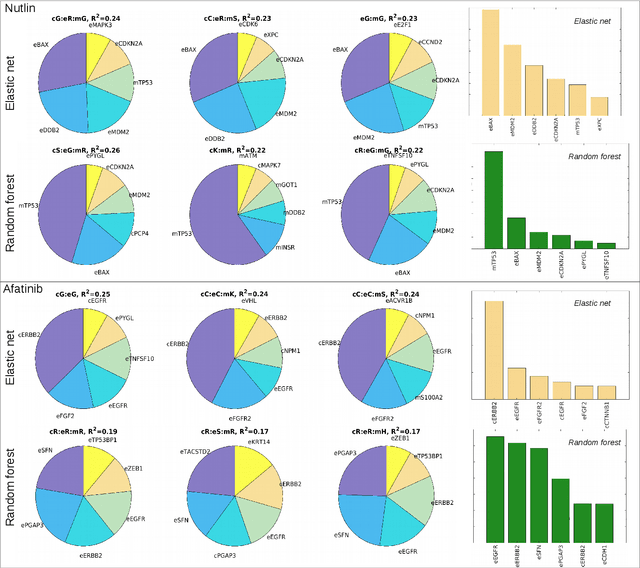
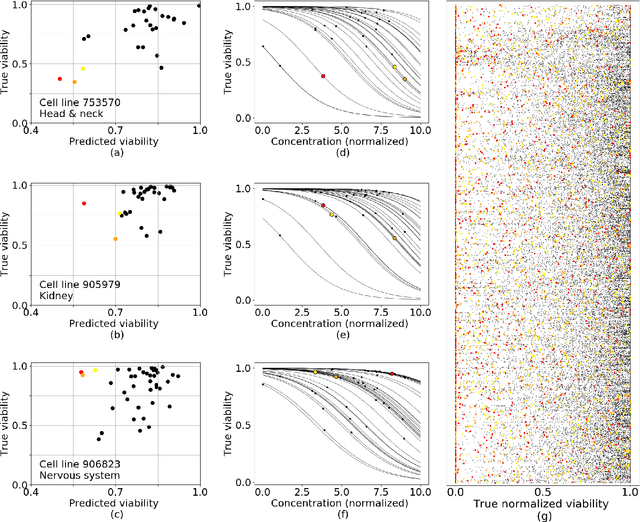
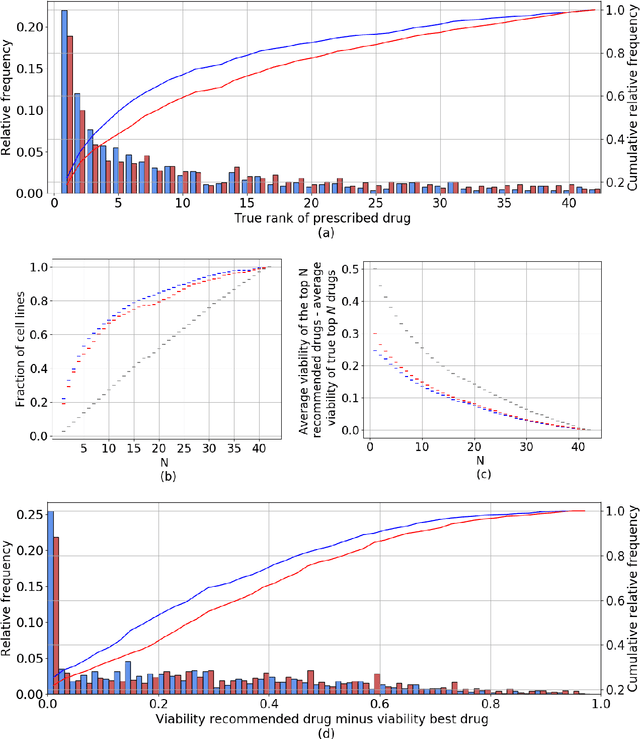
Personalizing drug prescriptions in cancer care based on genomic information requires associating genomic markers with treatment effects. This is an unsolved challenge requiring genomic patient data in yet unavailable volumes as well as appropriate quantitative methods. We attempt to solve this challenge for an experimental proxy for which sufficient data is available: 42 drugs tested on 1018 cancer cell lines. Our goal is to develop a method to identify the drug that is most promising based on a cell line's genomic information. For this, we need to identify for each drug the machine learning method, choice of hyperparameters and genomic features for optimal predictive performance. We extensively compare combinations of gene sets (both curated and random), genetic features, and machine learning algorithms for all 42 drugs. For each drug, the best performing combination (considering only the curated gene sets) is selected. We use these top model parameters for each drug to build and demonstrate a Drug Recommendation System (Dr.S). Insights resulting from this analysis are formulated as best practices for developing drug recommendation systems. The complete software system, called the Cell Line Analyzer, is written in Python and available on github.