 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLatent Chemical Space Searching for Plug-in Multi-objective Molecule Generation
Apr 10, 2024
Molecular generation, an essential method for identifying new drug structures, has been supported by advancements in machine learning and computational technology. However, challenges remain in multi-objective generation, model adaptability, and practical application in drug discovery. In this study, we developed a versatile 'plug-in' molecular generation model that incorporates multiple objectives related to target affinity, drug-likeness, and synthesizability, facilitating its application in various drug development contexts. We improved the Particle Swarm Optimization (PSO) in the context of drug discoveries, and identified PSO-ENP as the optimal variant for multi-objective molecular generation and optimization through comparative experiments. The model also incorporates a novel target-ligand affinity predictor, enhancing the model's utility by supporting three-dimensional information and improving synthetic feasibility. Case studies focused on generating and optimizing drug-like big marine natural products were performed, underscoring PSO-ENP's effectiveness and demonstrating its considerable potential for practical drug discovery applications.
Uni-RXN: A Unified Framework Bridging the Gap between Chemical Reaction Pretraining and Conditional Molecule Generation
Mar 14, 2023Chemical reactions are the fundamental building blocks of drug design and organic chemistry research. In recent years, there has been a growing need for a large-scale deep-learning framework that can efficiently capture the basic rules of chemical reactions. In this paper, we have proposed a unified framework that addresses both the reaction representation learning and molecule generation tasks, which allows for a more holistic approach. Inspired by the organic chemistry mechanism, we develop a novel pretraining framework that enables us to incorporate inductive biases into the model. Our framework achieves state-of-the-art results on challenging downstream tasks. By possessing chemical knowledge, this framework can be applied to reaction-based generative models, overcoming the limitations of current molecule generation models that rely on a small number of reaction templates. In the extensive experiments, our model generates synthesizable drug-like structures of high quality. Overall, our work presents a significant step toward a large-scale deep-learning framework for a variety of reaction-based applications.
TF3P: Three-dimensional Force Fields Fingerprint Learned by Deep Capsular Network
Dec 25, 2019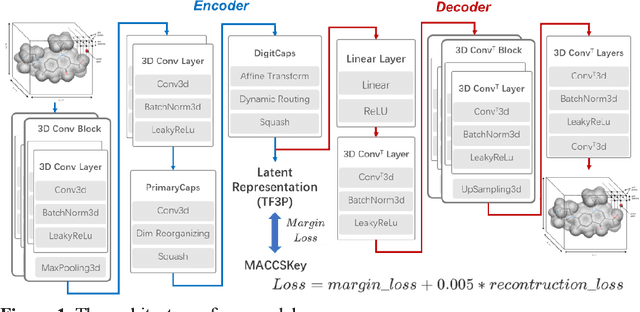
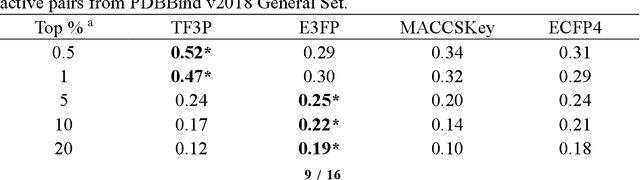
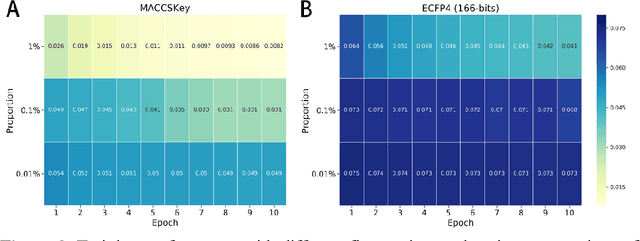
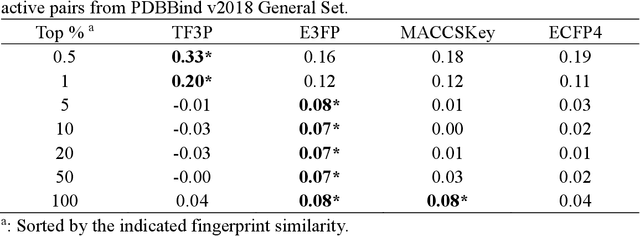
Molecular fingerprints are the workhorse in ligand-based drug discovery. In recent years, increasing number of research papers reported fascinating results on using deep neural networks to learn 2D molecular representations as fingerprints. One may anticipate that the integration of deep learning would also contribute to the prosperity of 3D fingerprints. Here, we presented a new 3D small molecule fingerprint, the three-dimensional force fields fingerprint (TF3P), learned by deep capsular network whose training is in no need of labeled dataset for specific predictive tasks. TF3P can encode the 3D force fields information of molecules and demonstrates its stronger ability to capture 3D structural changes, recognize molecules alike in 3D but not in 2D, and recognize similar targets inaccessible by other fingerprints, including the solely existing 3D fingerprint E3FP, based on only ligands similarity. Furthermore, TF3P is compatible with both statistical models (e.g. similarity ensemble approach) and machine learning models. Altogether, we report TF3P as a new 3D small molecule fingerprint with promising future in ligand-based drug discovery.
DeepScaffold: a comprehensive tool for scaffold-based de novo drug discovery using deep learning
Sep 05, 2019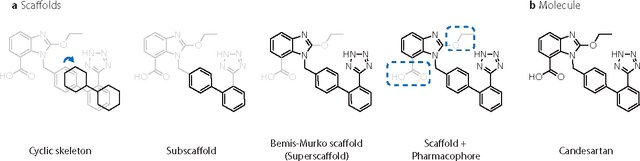

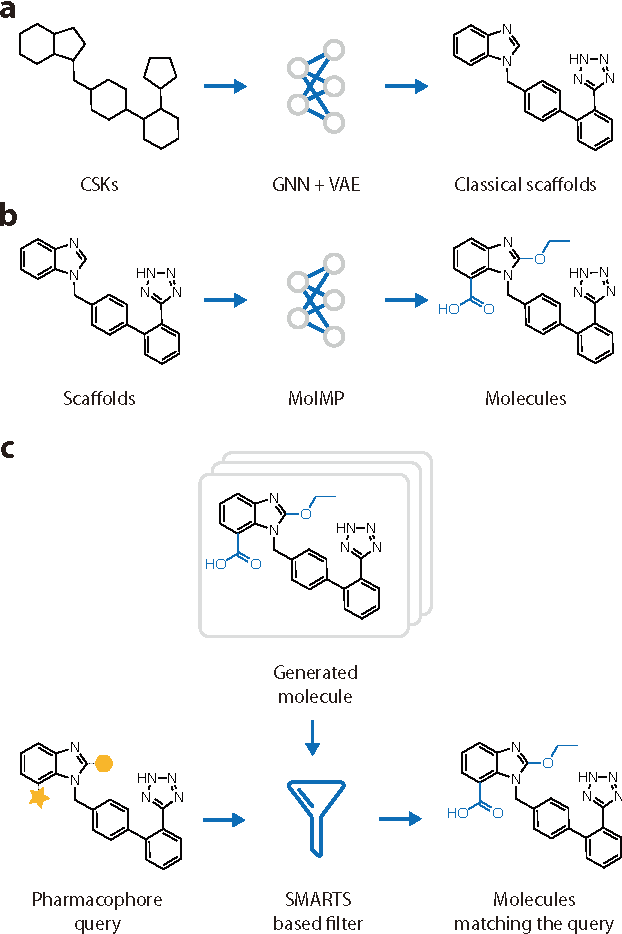
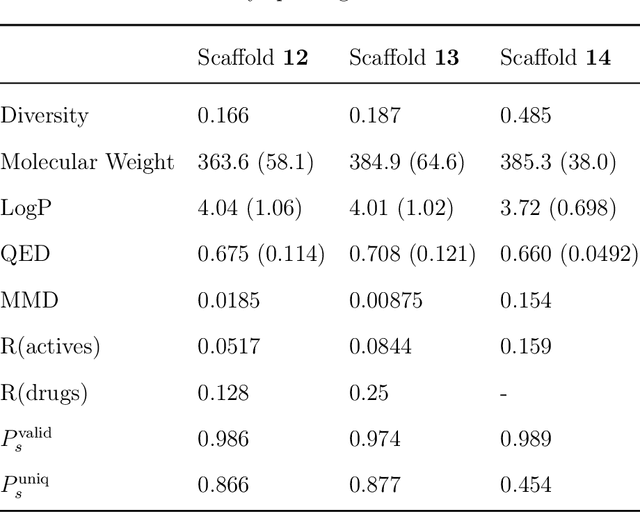
The ultimate goal of drug design is to find novel compounds with desirable pharmacological properties. Designing molecules retaining particular scaffolds as the core structures of the molecules is one of the efficient ways to obtain potential drug candidates with desirable properties. We proposed a scaffold-based molecular generative model for scaffold-based drug discovery, which performs molecule generation based on a wide spectrum of scaffold definitions, including BM-scaffolds, cyclic skeletons, as well as scaffolds with specifications on side-chain properties. The model can generalize the learned chemical rules of adding atoms and bonds to a given scaffold. Furthermore, the generated compounds were evaluated by molecular docking in DRD2 targets and the results demonstrated that this approach can be effectively applied to solve several drug design problems, including the generation of compounds containing a given scaffold and de novo drug design of potential drug candidates with specific docking scores. Finally, a command line interface is created.
Multi-Objective De Novo Drug Design with Conditional Graph Generative Model
Apr 21, 2018

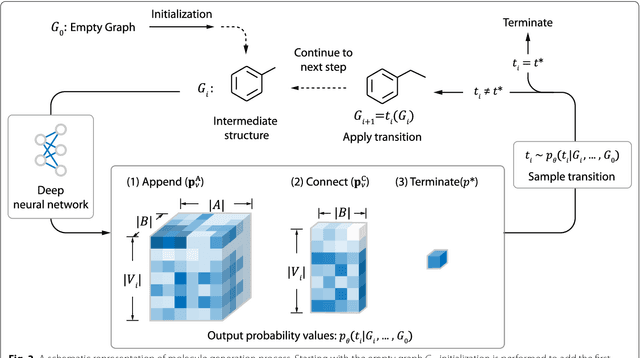
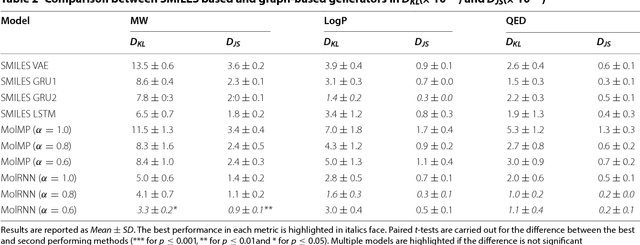
Recently, deep generative models have revealed itself as a promising way of performing de novo molecule design. However, previous research has focused mainly on generating SMILES strings instead of molecular graphs. Although current graph generative models are available, they are often too general and computationally expensive, which restricts their application to molecules with small sizes. In this work, a new de novo molecular design framework is proposed based on a type sequential graph generators that do not use atom level recurrent units. Compared with previous graph generative models, the proposed method is much more tuned for molecule generation and have been scaled up to cover significantly larger molecules in the ChEMBL database. It is shown that the graph-based model outperforms SMILES based models in a variety of metrics, especially in the rate of valid outputs. For the application of drug design tasks, conditional graph generative model is employed. This method offers higher flexibility compared to previous fine-tuning based approach and is suitable for generation based on multiple objectives. This approach is applied to solve several drug design problems, including the generation of compounds containing a given scaffold, generation of compounds with specific drug-likeness and synthetic accessibility requirements, as well as generating dual inhibitors against JNK3 and GSK3$\beta$. Results show high enrichment rates for outputs satisfying the given requirements.