 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTF3P: Three-dimensional Force Fields Fingerprint Learned by Deep Capsular Network
Dec 25, 2019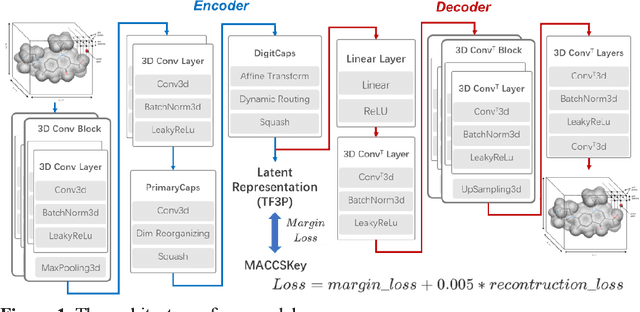
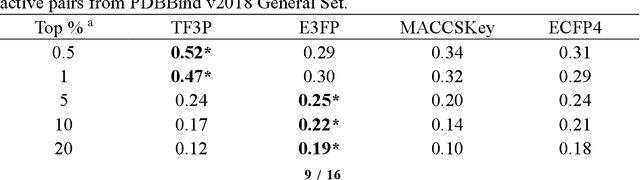
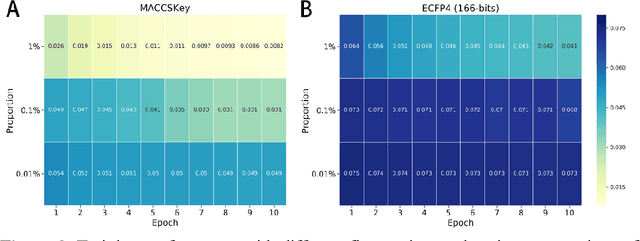
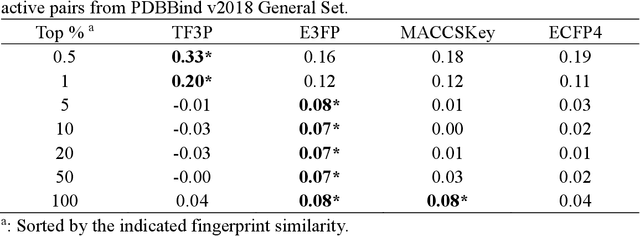
Molecular fingerprints are the workhorse in ligand-based drug discovery. In recent years, increasing number of research papers reported fascinating results on using deep neural networks to learn 2D molecular representations as fingerprints. One may anticipate that the integration of deep learning would also contribute to the prosperity of 3D fingerprints. Here, we presented a new 3D small molecule fingerprint, the three-dimensional force fields fingerprint (TF3P), learned by deep capsular network whose training is in no need of labeled dataset for specific predictive tasks. TF3P can encode the 3D force fields information of molecules and demonstrates its stronger ability to capture 3D structural changes, recognize molecules alike in 3D but not in 2D, and recognize similar targets inaccessible by other fingerprints, including the solely existing 3D fingerprint E3FP, based on only ligands similarity. Furthermore, TF3P is compatible with both statistical models (e.g. similarity ensemble approach) and machine learning models. Altogether, we report TF3P as a new 3D small molecule fingerprint with promising future in ligand-based drug discovery.
DeepScaffold: a comprehensive tool for scaffold-based de novo drug discovery using deep learning
Sep 05, 2019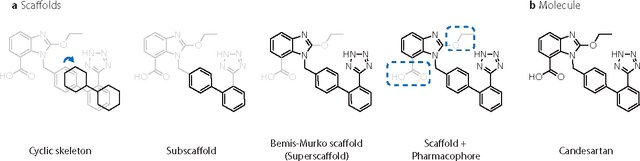

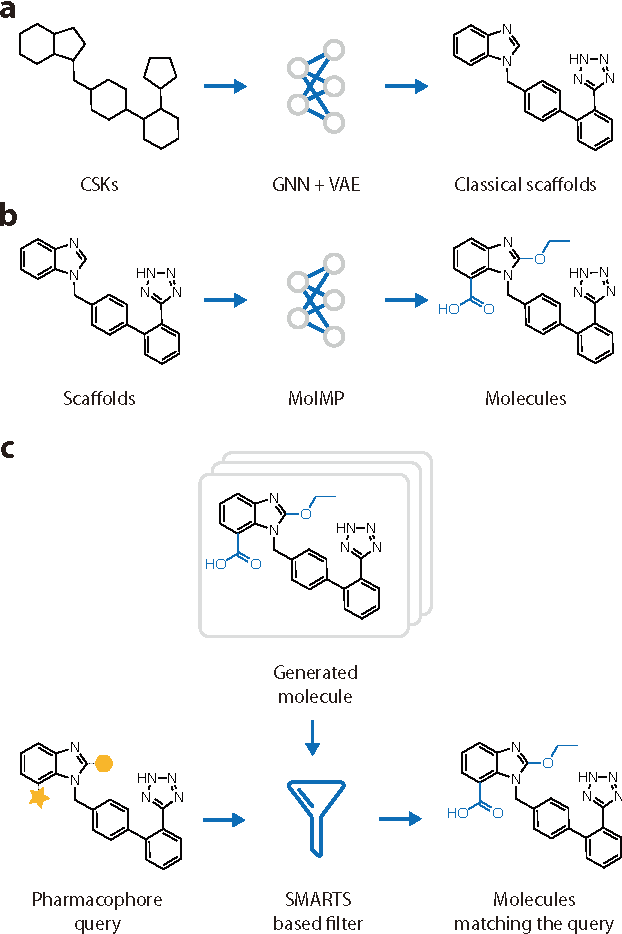
The ultimate goal of drug design is to find novel compounds with desirable pharmacological properties. Designing molecules retaining particular scaffolds as the core structures of the molecules is one of the efficient ways to obtain potential drug candidates with desirable properties. We proposed a scaffold-based molecular generative model for scaffold-based drug discovery, which performs molecule generation based on a wide spectrum of scaffold definitions, including BM-scaffolds, cyclic skeletons, as well as scaffolds with specifications on side-chain properties. The model can generalize the learned chemical rules of adding atoms and bonds to a given scaffold. Furthermore, the generated compounds were evaluated by molecular docking in DRD2 targets and the results demonstrated that this approach can be effectively applied to solve several drug design problems, including the generation of compounds containing a given scaffold and de novo drug design of potential drug candidates with specific docking scores. Finally, a command line interface is created.