 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLingo3DMol: Generation of a Pocket-based 3D Molecule using a Language Model
May 17, 2023Structure-based drug design powered by deep generative models have attracted increasing research interest in recent years. Language models have demonstrated a robust capacity for generating valid molecules in 2D structures, while methods based on geometric deep learning can directly produce molecules with accurate 3D coordinates. Inspired by both methods, this article proposes a pocket-based 3D molecule generation method that leverages the language model with the ability to generate 3D coordinates. High quality protein-ligand complex data are insufficient; hence, a perturbation and restoration pre-training task is designed that can utilize vast amounts of small-molecule data. A new molecular representation, a fragment-based SMILES with local and global coordinates, is also presented, enabling the language model to learn molecular topological structures and spatial position information effectively. Ultimately, CrossDocked and DUD-E dataset is employed for evaluation and additional metrics are introduced. This method achieves state-of-the-art performance in nearly all metrics, notably in terms of binding patterns, drug-like properties, rational conformations, and inference speed. Our model is available as an online service to academic users via sw3dmg.stonewise.cn
DeepScaffold: a comprehensive tool for scaffold-based de novo drug discovery using deep learning
Sep 05, 2019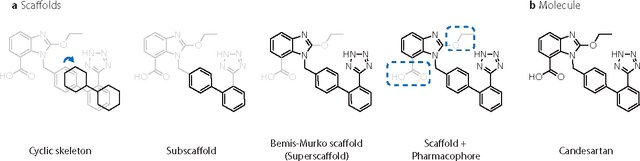

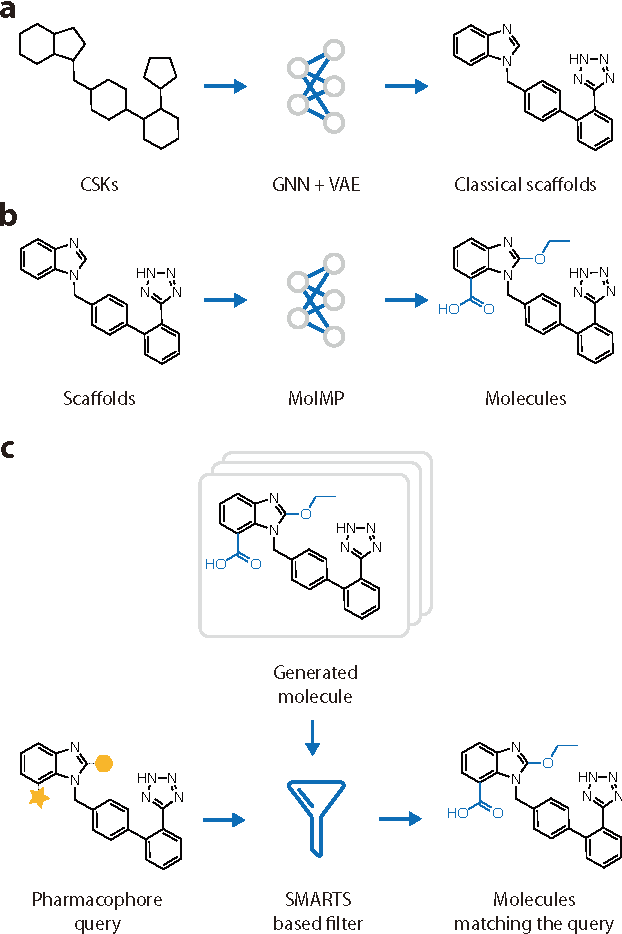
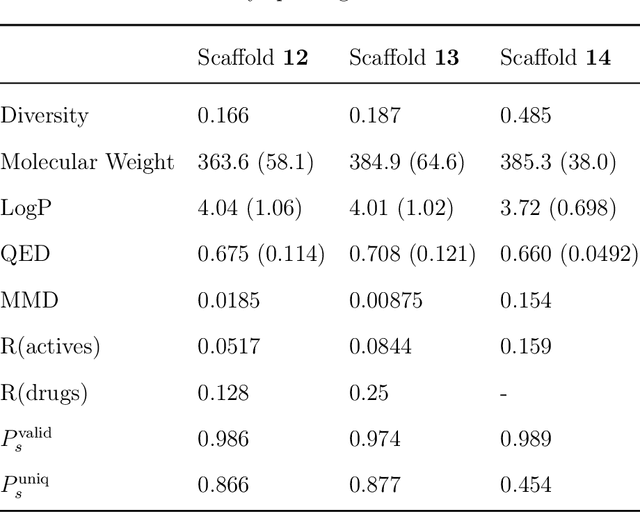
The ultimate goal of drug design is to find novel compounds with desirable pharmacological properties. Designing molecules retaining particular scaffolds as the core structures of the molecules is one of the efficient ways to obtain potential drug candidates with desirable properties. We proposed a scaffold-based molecular generative model for scaffold-based drug discovery, which performs molecule generation based on a wide spectrum of scaffold definitions, including BM-scaffolds, cyclic skeletons, as well as scaffolds with specifications on side-chain properties. The model can generalize the learned chemical rules of adding atoms and bonds to a given scaffold. Furthermore, the generated compounds were evaluated by molecular docking in DRD2 targets and the results demonstrated that this approach can be effectively applied to solve several drug design problems, including the generation of compounds containing a given scaffold and de novo drug design of potential drug candidates with specific docking scores. Finally, a command line interface is created.