 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSingle-Cell Clustering via Dual-Graph Alignment
Nov 28, 2023



In recent years, the field of single-cell RNA sequencing has seen a surge in the development of clustering methods. These methods enable the identification of cell subpopulations, thereby facilitating the understanding of tumor microenvironments. Despite their utility, most existing clustering algorithms primarily focus on the attribute information provided by the cell matrix or the network structure between cells, often neglecting the network between genes. This oversight could lead to loss of information and clustering results that lack clinical significance. To address this limitation, we develop an advanced single-cell clustering model incorporating dual-graph alignment, which integrates gene network information into the clustering process based on self-supervised and unsupervised optimization. Specifically, we designed a graph-based autoencoder enhanced by an attention mechanism to effectively capture relationships between cells. Moreover, we performed the node2vec method on Protein-Protein Interaction (PPI) networks to derive the gene network structure and maintained this structure throughout the clustering process. Our proposed method has been demonstrated to be effective through experimental results, showcasing its ability to optimize clustering outcomes while preserving the original associations between cells and genes. This research contributes to obtaining accurate cell subpopulations and generates clustering results that more closely resemble real-world biological scenarios. It provides better insights into the characteristics and distribution of diseased cells, ultimately building a foundation for early disease diagnosis and treatment.
Single-cell Multi-view Clustering via Community Detection with Unknown Number of Clusters
Nov 28, 2023



Single-cell multi-view clustering enables the exploration of cellular heterogeneity within the same cell from different views. Despite the development of several multi-view clustering methods, two primary challenges persist. Firstly, most existing methods treat the information from both single-cell RNA (scRNA) and single-cell Assay of Transposase Accessible Chromatin (scATAC) views as equally significant, overlooking the substantial disparity in data richness between the two views. This oversight frequently leads to a degradation in overall performance. Additionally, the majority of clustering methods necessitate manual specification of the number of clusters by users. However, for biologists dealing with cell data, precisely determining the number of distinct cell types poses a formidable challenge. To this end, we introduce scUNC, an innovative multi-view clustering approach tailored for single-cell data, which seamlessly integrates information from different views without the need for a predefined number of clusters. The scUNC method comprises several steps: initially, it employs a cross-view fusion network to create an effective embedding, which is then utilized to generate initial clusters via community detection. Subsequently, the clusters are automatically merged and optimized until no further clusters can be merged. We conducted a comprehensive evaluation of scUNC using three distinct single-cell datasets. The results underscored that scUNC outperforms the other baseline methods.
Dual-channel Prototype Network for few-shot Classification of Pathological Images
Nov 14, 2023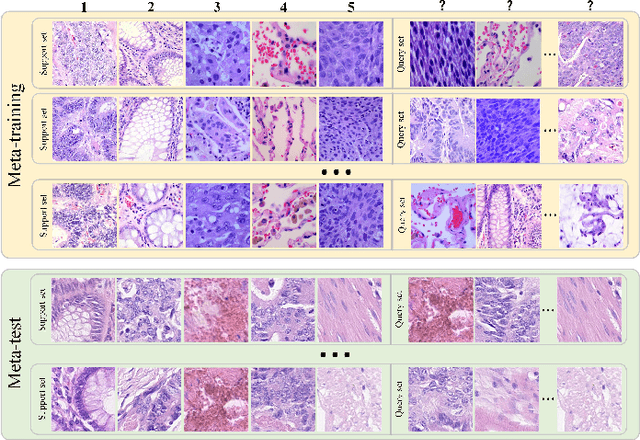
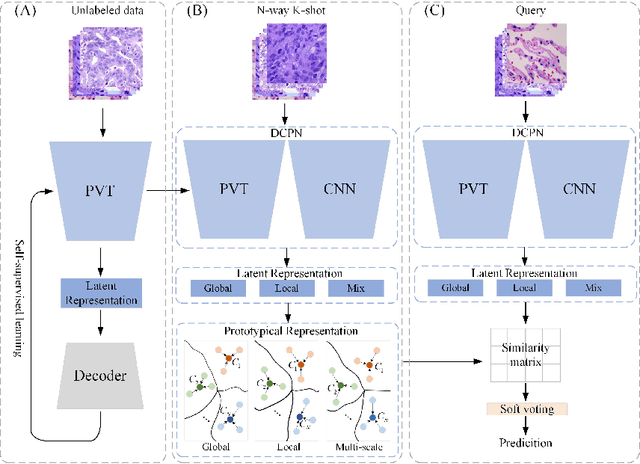
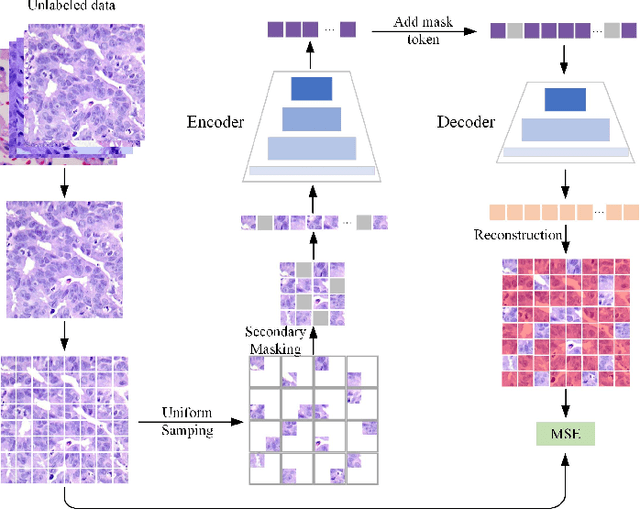
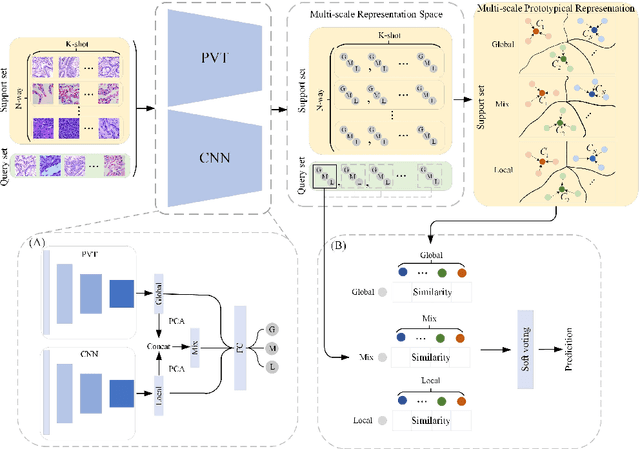
In pathology, the rarity of certain diseases and the complexity in annotating pathological images significantly hinder the creation of extensive, high-quality datasets. This limitation impedes the progress of deep learning-assisted diagnostic systems in pathology. Consequently, it becomes imperative to devise a technology that can discern new disease categories from a minimal number of annotated examples. Such a technology would substantially advance deep learning models for rare diseases. Addressing this need, we introduce the Dual-channel Prototype Network (DCPN), rooted in the few-shot learning paradigm, to tackle the challenge of classifying pathological images with limited samples. DCPN augments the Pyramid Vision Transformer (PVT) framework for few-shot classification via self-supervised learning and integrates it with convolutional neural networks. This combination forms a dual-channel architecture that extracts multi-scale, highly precise pathological features. The approach enhances the versatility of prototype representations and elevates the efficacy of prototype networks in few-shot pathological image classification tasks. We evaluated DCPN using three publicly available pathological datasets, configuring small-sample classification tasks that mirror varying degrees of clinical scenario domain shifts. Our experimental findings robustly affirm DCPN's superiority in few-shot pathological image classification, particularly in tasks within the same domain, where it achieves the benchmarks of supervised learning.
TMac: Temporal Multi-Modal Graph Learning for Acoustic Event Classification
Sep 26, 2023Audiovisual data is everywhere in this digital age, which raises higher requirements for the deep learning models developed on them. To well handle the information of the multi-modal data is the key to a better audiovisual modal. We observe that these audiovisual data naturally have temporal attributes, such as the time information for each frame in the video. More concretely, such data is inherently multi-modal according to both audio and visual cues, which proceed in a strict chronological order. It indicates that temporal information is important in multi-modal acoustic event modeling for both intra- and inter-modal. However, existing methods deal with each modal feature independently and simply fuse them together, which neglects the mining of temporal relation and thus leads to sub-optimal performance. With this motivation, we propose a Temporal Multi-modal graph learning method for Acoustic event Classification, called TMac, by modeling such temporal information via graph learning techniques. In particular, we construct a temporal graph for each acoustic event, dividing its audio data and video data into multiple segments. Each segment can be considered as a node, and the temporal relationships between nodes can be considered as timestamps on their edges. In this case, we can smoothly capture the dynamic information in intra-modal and inter-modal. Several experiments are conducted to demonstrate TMac outperforms other SOTA models in performance. Our code is available at https://github.com/MGitHubL/TMac.