 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAtom-by-atom protein generation and beyond with language models
Aug 16, 2023
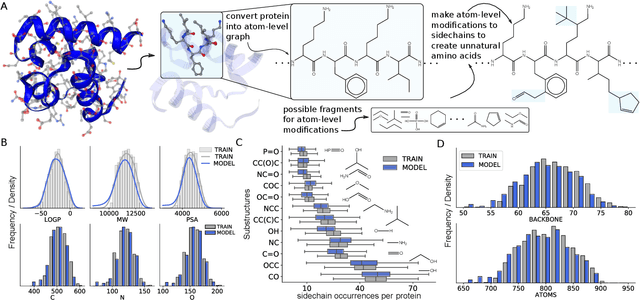


Protein language models learn powerful representations directly from sequences of amino acids. However, they are constrained to generate proteins with only the set of amino acids represented in their vocabulary. In contrast, chemical language models learn atom-level representations of smaller molecules that include every atom, bond, and ring. In this work, we show that chemical language models can learn atom-level representations of proteins enabling protein generation unconstrained to the standard genetic code and far beyond it. In doing so, we show that language models can generate entire proteins atom by atom -- effectively learning the multiple hierarchical layers of molecular information that define proteins from their primary sequence to their secondary, and tertiary structure. We demonstrate language models are able to explore beyond protein space -- generating proteins with modified sidechains that form unnatural amino acids. Even further, we find that language models can explore chemical space and protein space simultaneously and generate novel examples of protein-drug conjugates. The results demonstrate the potential for biomolecular design at the atom level using language models.
Language models can generate molecules, materials, and protein binding sites directly in three dimensions as XYZ, CIF, and PDB files
May 09, 2023Language models are powerful tools for molecular design. Currently, the dominant paradigm is to parse molecular graphs into linear string representations that can easily be trained on. This approach has been very successful, however, it is limited to chemical structures that can be completely represented by a graph -- like organic molecules -- while materials and biomolecular structures like protein binding sites require a more complete representation that includes the relative positioning of their atoms in space. In this work, we show how language models, without any architecture modifications, trained using next-token prediction -- can generate novel and valid structures in three dimensions from various substantially different distributions of chemical structures. In particular, we demonstrate that language models trained directly on sequences derived directly from chemical file formats like XYZ files, Crystallographic Information files (CIFs), or Protein Data Bank files (PDBs) can directly generate molecules, crystals, and protein binding sites in three dimensions. Furthermore, despite being trained on chemical file sequences -- language models still achieve performance comparable to state-of-the-art models that use graph and graph-derived string representations, as well as other domain-specific 3D generative models. In doing so, we demonstrate that it is not necessary to use simplified molecular representations to train chemical language models -- that they are powerful generative models capable of directly exploring chemical space in three dimensions for very different structures.
Scalable Fragment-Based 3D Molecular Design with Reinforcement Learning
Feb 01, 2022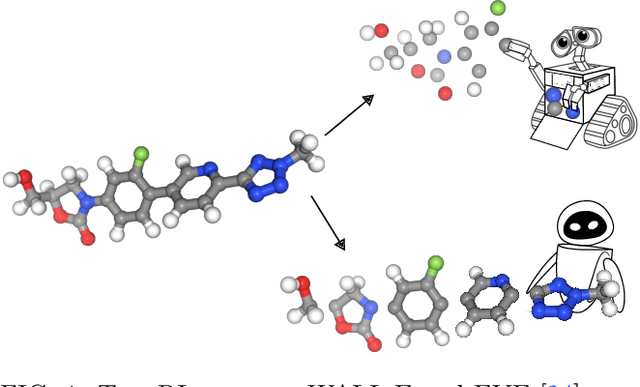
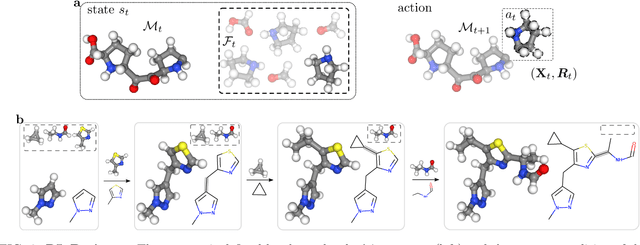
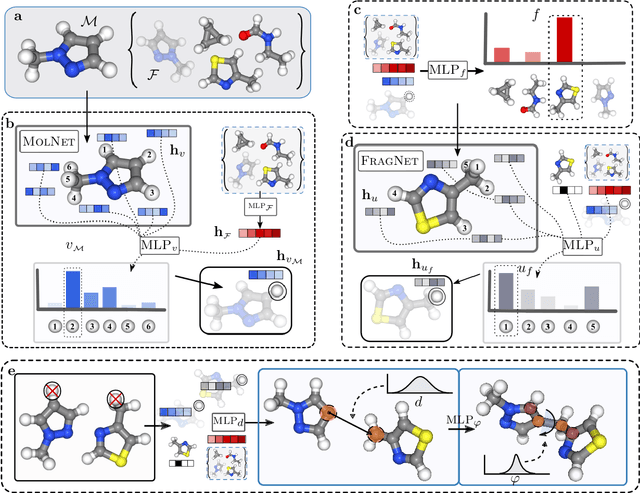
Machine learning has the potential to automate molecular design and drastically accelerate the discovery of new functional compounds. Towards this goal, generative models and reinforcement learning (RL) using string and graph representations have been successfully used to search for novel molecules. However, these approaches are limited since their representations ignore the three-dimensional (3D) structure of molecules. In fact, geometry plays an important role in many applications in inverse molecular design, especially in drug discovery. Thus, it is important to build models that can generate molecular structures in 3D space based on property-oriented geometric constraints. To address this, one approach is to generate molecules as 3D point clouds by sequentially placing atoms at locations in space -- this allows the process to be guided by physical quantities such as energy or other properties. However, this approach is inefficient as placing individual atoms makes the exploration unnecessarily deep, limiting the complexity of molecules that can be generated. Moreover, when optimizing a molecule, organic and medicinal chemists use known fragments and functional groups, not single atoms. We introduce a novel RL framework for scalable 3D design that uses a hierarchical agent to build molecules by placing molecular substructures sequentially in 3D space, thus attempting to build on the existing human knowledge in the field of molecular design. In a variety of experiments with different substructures, we show that our agent, guided only by energy considerations, can efficiently learn to produce molecules with over 100 atoms from many distributions including drug-like molecules, organic LED molecules, and biomolecules.
Keeping it Simple: Language Models can learn Complex Molecular Distributions
Dec 06, 2021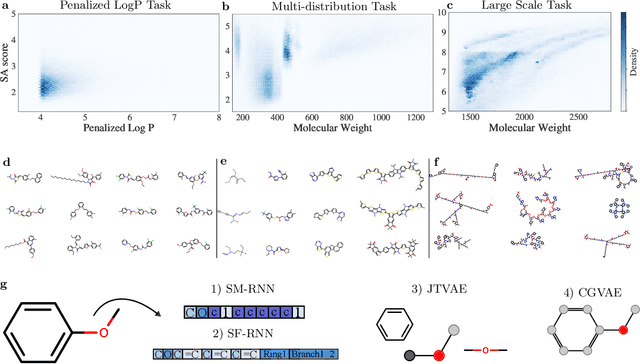
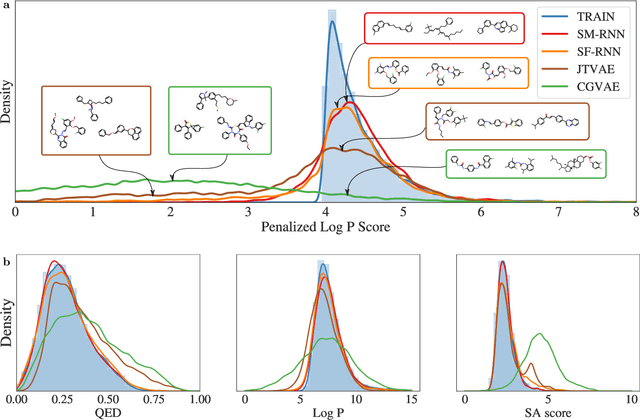
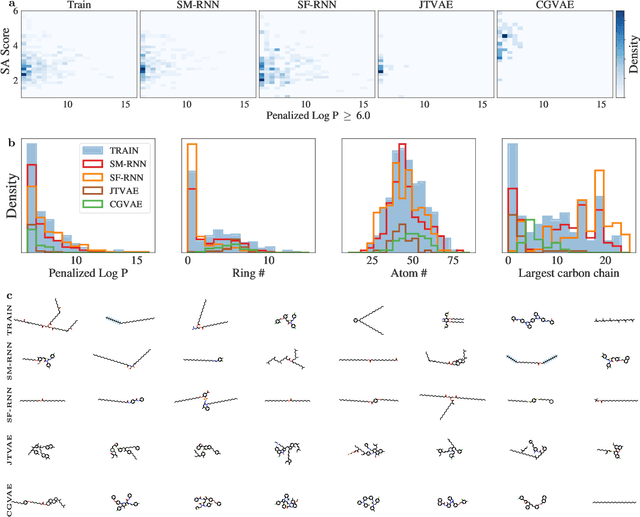
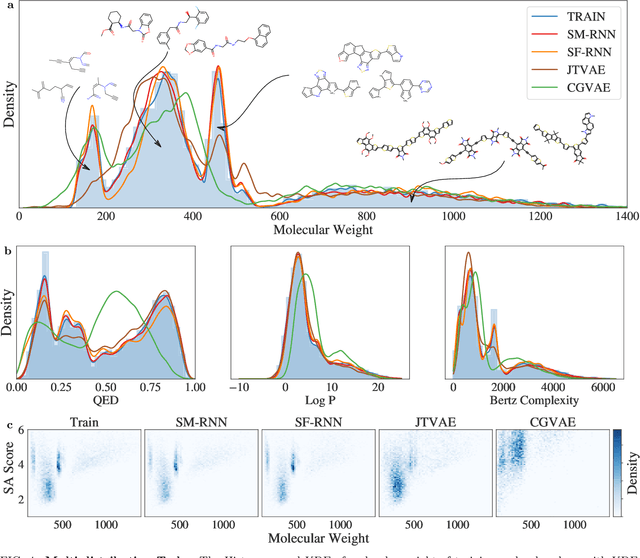
Deep generative models of molecules have grown immensely in popularity, trained on relevant datasets, these models are used to search through chemical space. The downstream utility of generative models for the inverse design of novel functional compounds depends on their ability to learn a training distribution of molecules. The most simple example is a language model that takes the form of a recurrent neural network and generates molecules using a string representation. More sophisticated are graph generative models, which sequentially construct molecular graphs and typically achieve state of the art results. However, recent work has shown that language models are more capable than once thought, particularly in the low data regime. In this work, we investigate the capacity of simple language models to learn distributions of molecules. For this purpose, we introduce several challenging generative modeling tasks by compiling especially complex distributions of molecules. On each task, we evaluate the ability of language models as compared with two widely used graph generative models. The results demonstrate that language models are powerful generative models, capable of adeptly learning complex molecular distributions -- and yield better performance than the graph models. Language models can accurately generate: distributions of the highest scoring penalized LogP molecules in ZINC15, multi-modal molecular distributions as well as the largest molecules in PubChem.
Learning quantum dynamics with latent neural ODEs
Oct 20, 2021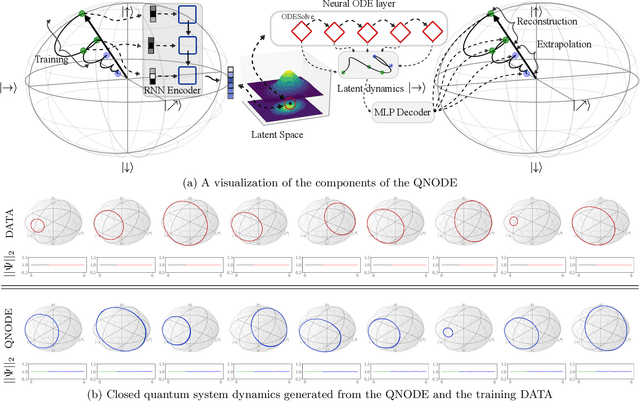
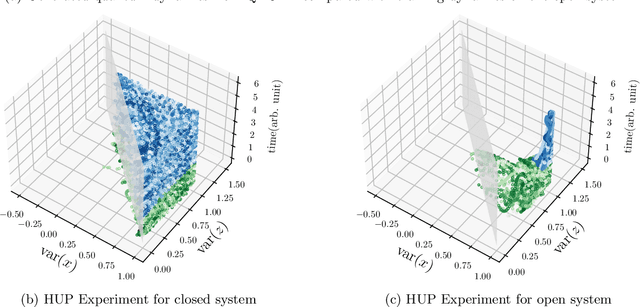
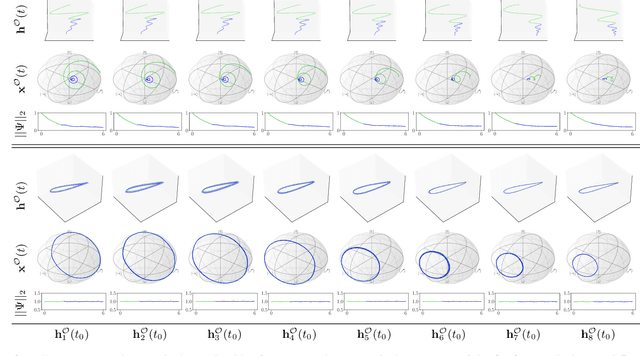
The core objective of machine-assisted scientific discovery is to learn physical laws from experimental data without prior knowledge of the systems in question. In the area of quantum physics, making progress towards these goals is significantly more challenging due to the curse of dimensionality as well as the counter-intuitive nature of quantum mechanics. Here, we present the QNODE, a latent neural ODE trained on dynamics from closed and open quantum systems. The QNODE can learn to generate quantum dynamics and extrapolate outside of its training region that satisfy the von Neumann and time-local Lindblad master equations for closed and open quantum systems. Furthermore the QNODE rediscovers quantum mechanical laws such as Heisenberg's uncertainty principle in a totally data-driven way, without constraints or guidance. Additionally, we show that trajectories that are generated from the QNODE and are close in its latent space have similar quantum dynamics while preserving the physics of the training system.
Learning Interpretable Representations of Entanglement in Quantum Optics Experiments using Deep Generative Models
Sep 06, 2021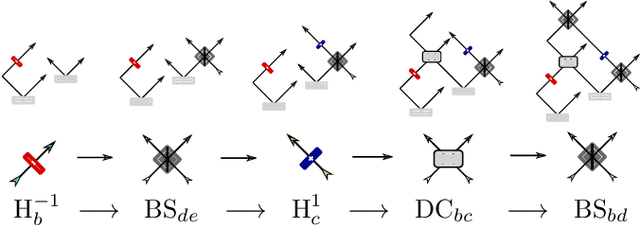
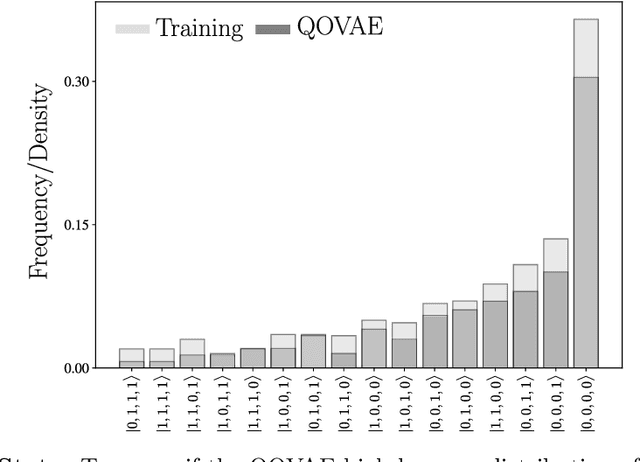
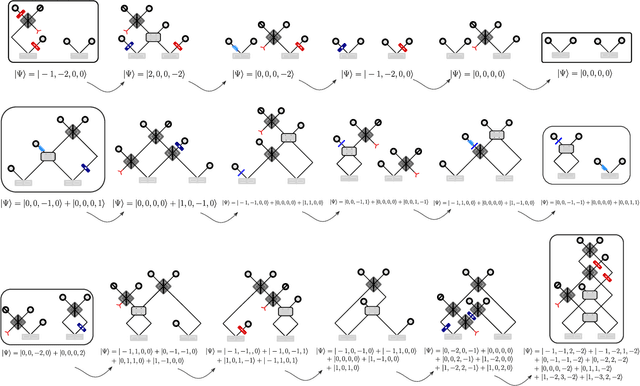
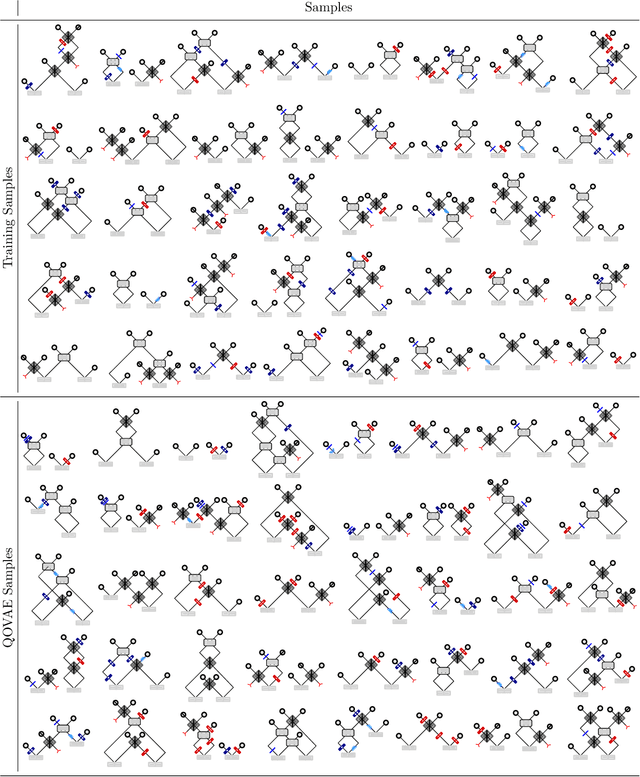
Quantum physics experiments produce interesting phenomena such as interference or entanglement, which is a core property of numerous future quantum technologies. The complex relationship between a quantum experiment's structure and its entanglement properties is essential to fundamental research in quantum optics but is difficult to intuitively understand. We present the first deep generative model of quantum optics experiments where a variational autoencoder (QOVAE) is trained on a dataset of experimental setups. In a series of computational experiments, we investigate the learned representation of the QOVAE and its internal understanding of the quantum optics world. We demonstrate that the QOVAE learns an intrepretable representation of quantum optics experiments and the relationship between experiment structure and entanglement. We show the QOVAE is able to generate novel experiments for highly entangled quantum states with specific distributions that match its training data. Importantly, we are able to fully interpret how the QOVAE structures its latent space, finding curious patterns that we can entirely explain in terms of quantum physics. The results demonstrate how we can successfully use and understand the internal representations of deep generative models in a complex scientific domain. The QOVAE and the insights from our investigations can be immediately applied to other physical systems throughout fundamental scientific research.
Bayesian Variational Optimization for Combinatorial Spaces
Nov 03, 2020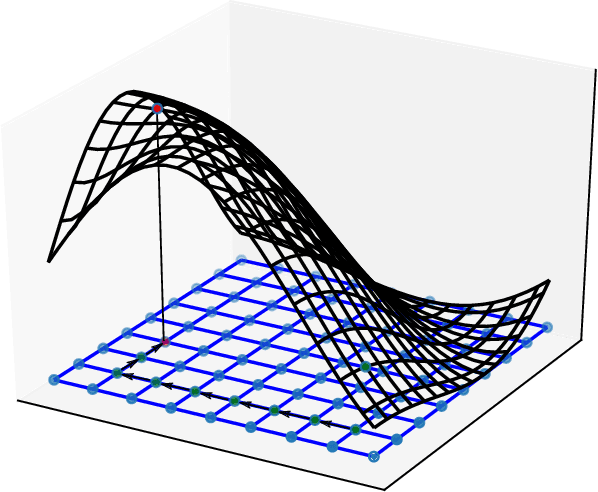
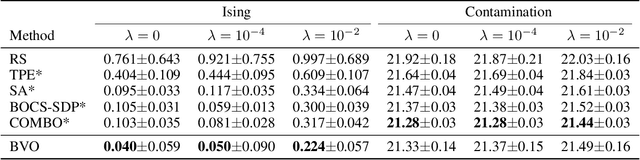
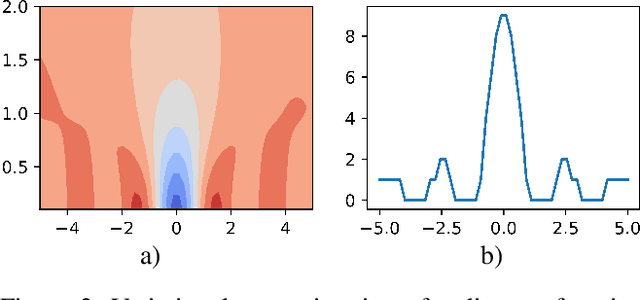
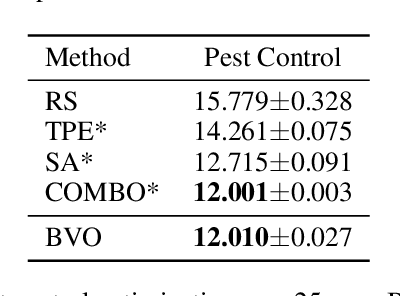
This paper focuses on Bayesian Optimization in combinatorial spaces. In many applications in the natural science. Broad applications include the study of molecules, proteins, DNA, device structures and quantum circuit designs, a on optimization over combinatorial categorical spaces is needed to find optimal or pareto-optimal solutions. However, only a limited amount of methods have been proposed to tackle this problem. Many of them depend on employing Gaussian Process for combinatorial Bayesian Optimizations. Gaussian Processes suffer from scalability issues for large data sizes as their scaling is cubic with respect to the number of data points. This is often impractical for optimizing large search spaces. Here, we introduce a variational Bayesian optimization method that combines variational optimization and continuous relaxations to the optimization of the acquisition function for Bayesian optimization. Critically, this method allows for gradient-based optimization and has the capability of optimizing problems with large data size and data dimensions. We have shown the performance of our method is comparable to state-of-the-art methods while maintaining its scalability advantages. We also applied our method in molecular optimization.
Neural Message Passing on High Order Paths
Feb 24, 2020

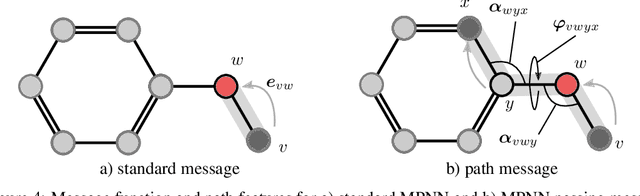
Graph neural network have achieved impressive results in predicting molecular properties, but they do not directly account for local and hidden structures in the graph such as functional groups and molecular geometry. At each propagation step, GNNs aggregate only over first order neighbours, ignoring important information contained in subsequent neighbours as well as the relationships between those higher order connections. In this work, we generalize graph neural nets to pass messages and aggregate across higher order paths. This allows for information to propagate over various levels and substructures of the graph. We demonstrate our model on a few tasks in molecular property prediction.
Graph Deconvolutional Generation
Feb 14, 2020
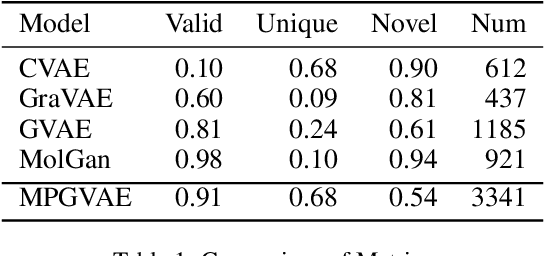
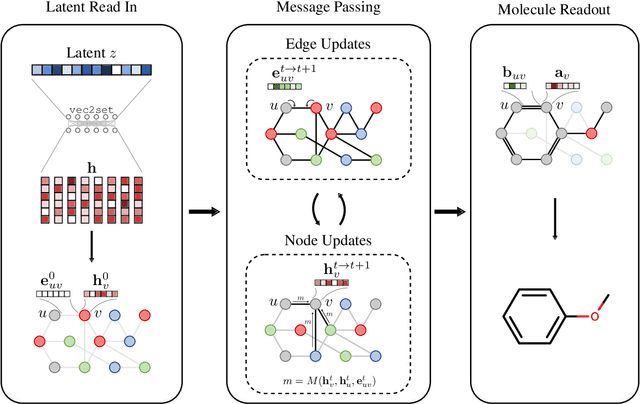
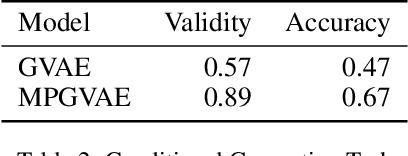
Graph generation is an extremely important task, as graphs are found throughout different areas of science and engineering. In this work, we focus on the modern equivalent of the Erdos-Renyi random graph model: the graph variational autoencoder (GVAE). This model assumes edges and nodes are independent in order to generate entire graphs at a time using a multi-layer perceptron decoder. As a result of these assumptions, GVAE has difficulty matching the training distribution and relies on an expensive graph matching procedure. We improve this class of models by building a message passing neural network into GVAE's encoder and decoder. We demonstrate our model on the specific task of generating small organic molecules