 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOptimizing Graph Transformer Networks with Graph-based Techniques
Jun 16, 2021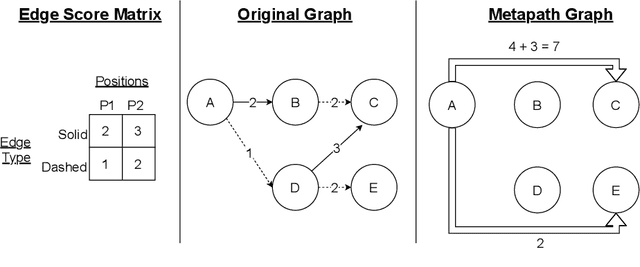
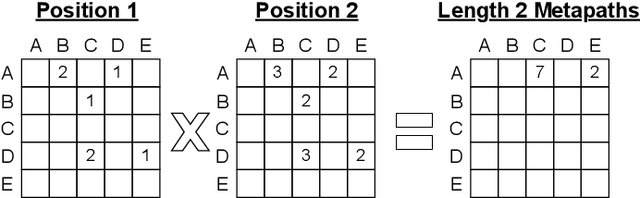
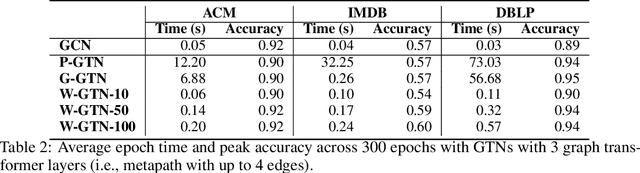
Graph transformer networks (GTN) are a variant of graph convolutional networks (GCN) that are targeted to heterogeneous graphs in which nodes and edges have associated type information that can be exploited to improve inference accuracy. GTNs learn important metapaths in the graph, create weighted edges for these metapaths, and use the resulting graph in a GCN. Currently, the only available implementation of GTNs uses dense matrix multiplication to find metapaths. Unfortunately, the space overhead of this approach can be large, so in practice it is used only for small graphs. In addition, the matrix-based implementation is not fine-grained enough to use random-walk based methods to optimize metapath finding. In this paper, we present a graph-based formulation and implementation of the GTN metapath finding problem. This graph-based formulation has two advantages over the matrix-based approach. First, it is more space efficient than the original GTN implementation and more compute-efficient for metapath sizes of practical interest. Second, it permits us to implement a sampling method that reduces the number of metapaths that must be enumerated, allowing the implementation to be used for larger graphs and larger metapath sizes. Experimental results show that our implementation is $6.5\times$ faster than the original GTN implementation on average for a metapath length of 4, and our sampling implementation is $155\times$ faster on average than this implementation without compromising on the accuracy of the GTN.
Biomedical Knowledge Graph Refinement and Completion using Graph Representation Learning and Top-K Similarity Measure
Dec 18, 2020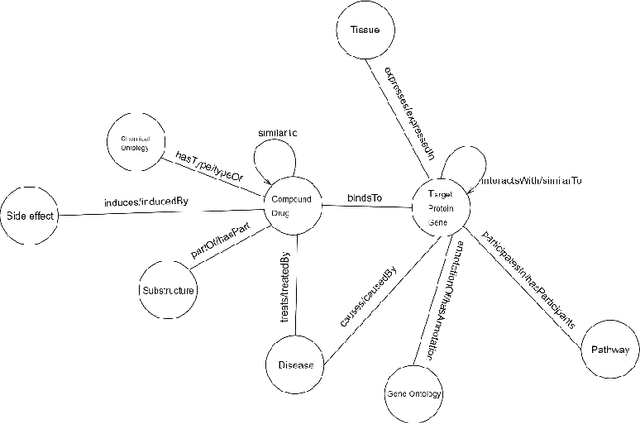

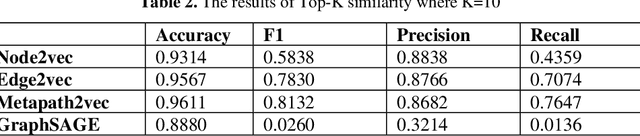
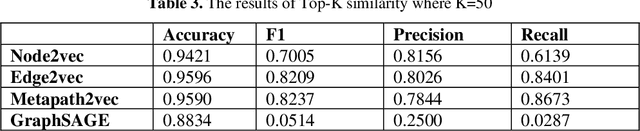
Knowledge Graphs have been one of the fundamental methods for integrating heterogeneous data sources. Integrating heterogeneous data sources is crucial, especially in the biomedical domain, where central data-driven tasks such as drug discovery rely on incorporating information from different biomedical databases. These databases contain various biological entities and relations such as proteins (PDB), genes (Gene Ontology), drugs (DrugBank), diseases (DDB), and protein-protein interactions (BioGRID). The process of semantically integrating heterogeneous biomedical databases is often riddled with imperfections. The quality of data-driven drug discovery relies on the accuracy of the mining methods used and the data's quality as well. Thus, having complete and refined biomedical knowledge graphs is central to achieving more accurate drug discovery outcomes. Here we propose using the latest graph representation learning and embedding models to refine and complete biomedical knowledge graphs. This preliminary work demonstrates learning discrete representations of the integrated biomedical knowledge graph Chem2Bio2RD [3]. We perform a knowledge graph completion and refinement task using a simple top-K cosine similarity measure between the learned embedding vectors to predict missing links between drugs and targets present in the data. We show that this simple procedure can be used alternatively to binary classifiers in link prediction.