 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDirect Molecular Conformation Generation
Paper and Code


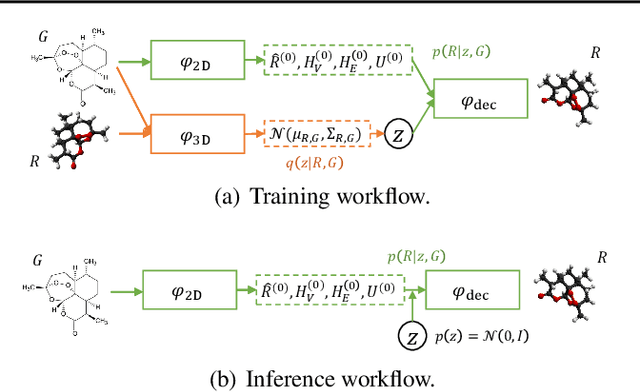

Molecular conformation generation aims to generate three-dimensional coordinates of all the atoms in a molecule and is an important task in bioinformatics and pharmacology. Previous distance-based methods first predict interatomic distances and then generate conformations based on them, which could result in conflicting distances. In this work, we propose a method that directly predicts the coordinates of atoms. We design a dedicated loss function for conformation generation, which is invariant to roto-translation of coordinates of conformations and permutation of symmetric atoms in molecules. We further design a backbone model that stacks multiple blocks, where each block refines the conformation generated by its preceding block. Our method achieves state-of-the-art results on four public benchmarks: on small-scale GEOM-QM9 and GEOM-Drugs which have $200$K training data, we can improve the previous best matching score by $3.5\%$ and $28.9\%$; on large-scale GEOM-QM9 and GEOM-Drugs which have millions of training data, those two improvements are $47.1\%$ and $36.3\%$. This shows the effectiveness of our method and the great potential of the direct approach. Our code is released at \url{https://github.com/DirectMolecularConfGen/DMCG}.