 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSelf-supervised denoising of visual field data improves detection of glaucoma progression
Nov 19, 2024Perimetric measurements provide insight into a patient's peripheral vision and day-to-day functioning and are the main outcome measure for identifying progression of visual damage from glaucoma. However, visual field data can be noisy, exhibiting high variance, especially with increasing damage. In this study, we demonstrate the utility of self-supervised deep learning in denoising visual field data from over 4000 patients to enhance its signal-to-noise ratio and its ability to detect true glaucoma progression. We deployed both a variational autoencoder (VAE) and a masked autoencoder to determine which self-supervised model best smooths the visual field data while reconstructing salient features that are less noisy and more predictive of worsening disease. Our results indicate that including a categorical p-value at every visual field location improves the smoothing of visual field data. Masked autoencoders led to cleaner denoised data than previous methods, such as variational autoencoders. A 4.7% increase in detection of progressing eyes with pointwise linear regression (PLR) was observed. The masked and variational autoencoders' smoothed data predicted glaucoma progression 2.3 months earlier when p-values were included compared to when they were not. The faster prediction of time to progression (TTP) and the higher percentage progression detected support our hypothesis that masking out visual field elements during training while including p-values at each location would improve the task of detection of visual field progression. Our study has clinically relevant implications regarding masking when training neural networks to denoise visual field data, resulting in earlier and more accurate detection of glaucoma progression. This denoising model can be integrated into future models for visual field analysis to enhance detection of glaucoma progression.
U-learning for Prediction Inference via Combinatory Multi-Subsampling: With Applications to LASSO and Neural Networks
Jul 22, 2024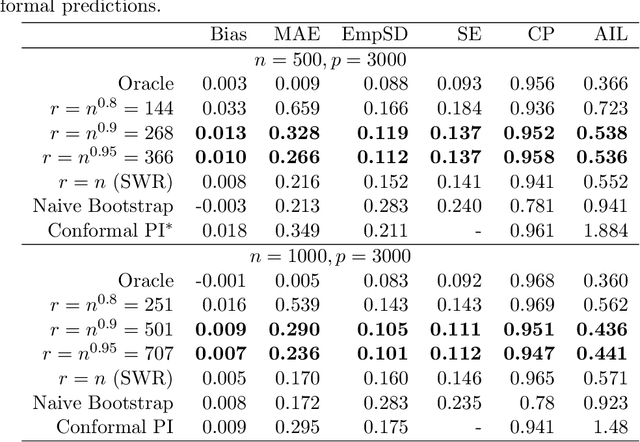
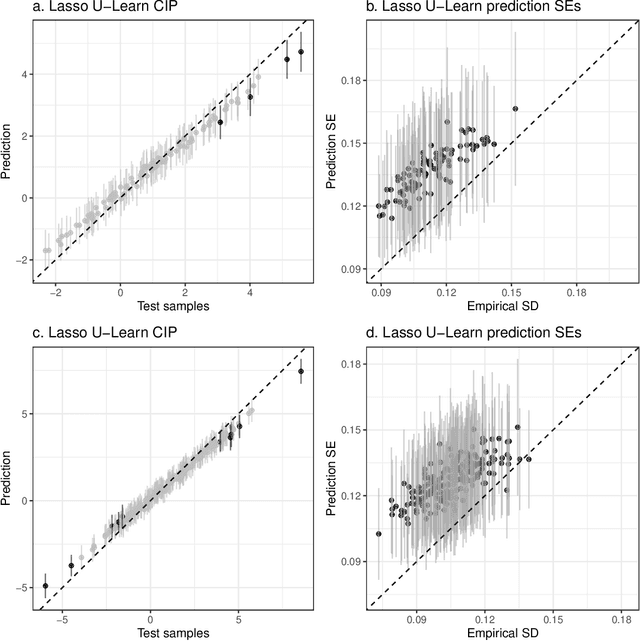
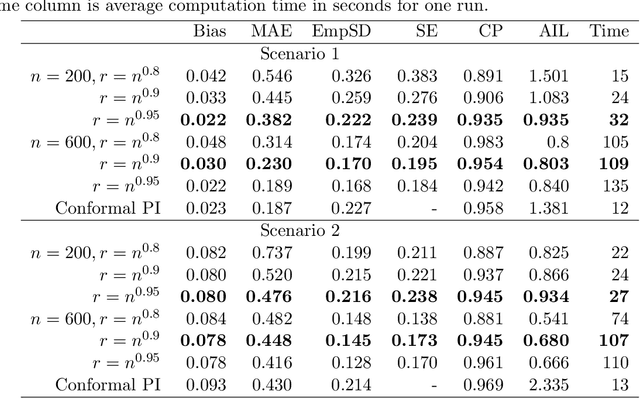
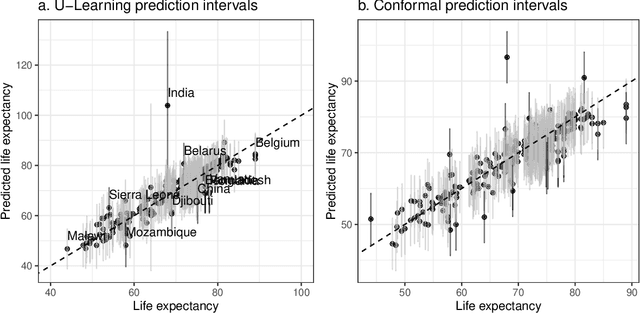
Epigenetic aging clocks play a pivotal role in estimating an individual's biological age through the examination of DNA methylation patterns at numerous CpG (Cytosine-phosphate-Guanine) sites within their genome. However, making valid inferences on predicted epigenetic ages, or more broadly, on predictions derived from high-dimensional inputs, presents challenges. We introduce a novel U-learning approach via combinatory multi-subsampling for making ensemble predictions and constructing confidence intervals for predictions of continuous outcomes when traditional asymptotic methods are not applicable. More specifically, our approach conceptualizes the ensemble estimators within the framework of generalized U-statistics and invokes the H\'ajek projection for deriving the variances of predictions and constructing confidence intervals with valid conditional coverage probabilities. We apply our approach to two commonly used predictive algorithms, Lasso and deep neural networks (DNNs), and illustrate the validity of inferences with extensive numerical studies. We have applied these methods to predict the DNA methylation age (DNAmAge) of patients with various health conditions, aiming to accurately characterize the aging process and potentially guide anti-aging interventions.
An Overview of Healthcare Data Analytics With Applications to the COVID-19 Pandemic
Nov 25, 2021
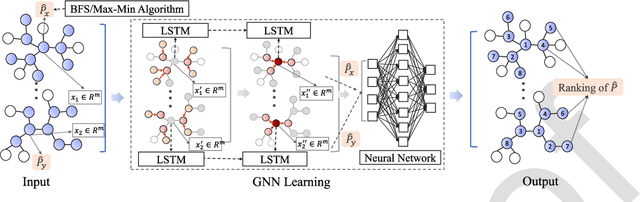

In the era of big data, standard analysis tools may be inadequate for making inference and there is a growing need for more efficient and innovative ways to collect, process, analyze and interpret the massive and complex data. We provide an overview of challenges in big data problems and describe how innovative analytical methods, machine learning tools and metaheuristics can tackle general healthcare problems with a focus on the current pandemic. In particular, we give applications of modern digital technology, statistical methods, data platforms and data integration systems to improve diagnosis and treatment of diseases in clinical research and novel epidemiologic tools to tackle infection source problems, such as finding Patient Zero in the spread of epidemics. We make the case that analyzing and interpreting big data is a very challenging task that requires a multi-disciplinary effort to continuously create more effective methodologies and powerful tools to transfer data information into knowledge that enables informed decision making.
Inference for High Dimensional Censored Quantile Regression
Jul 22, 2021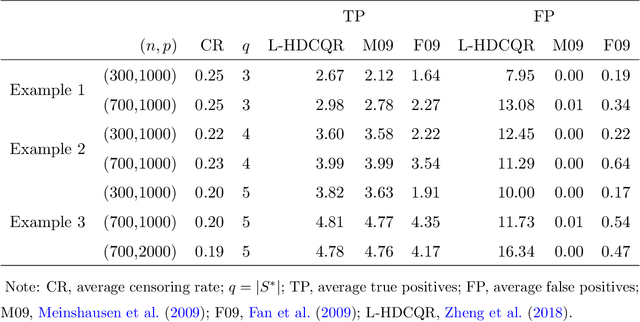
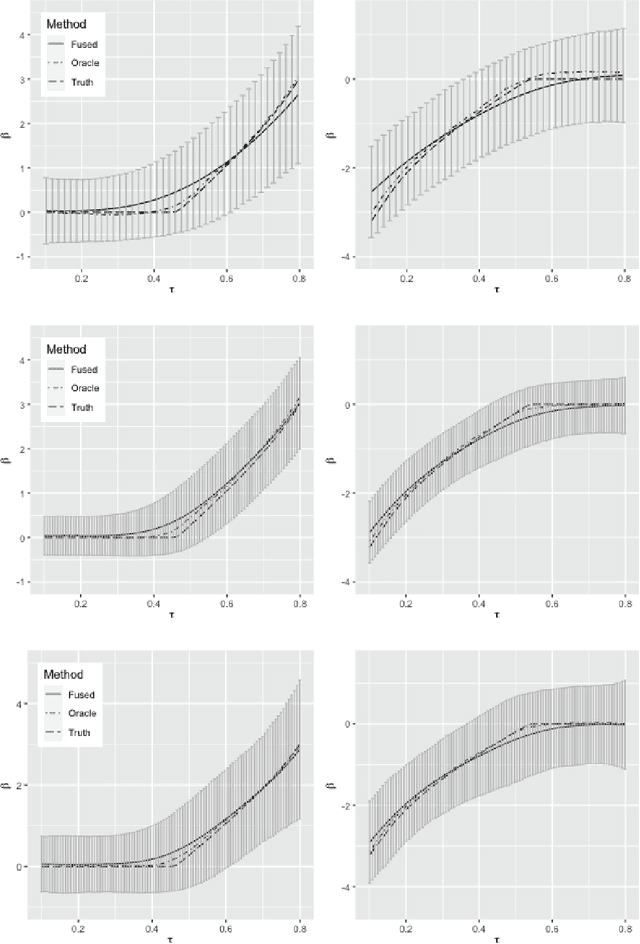

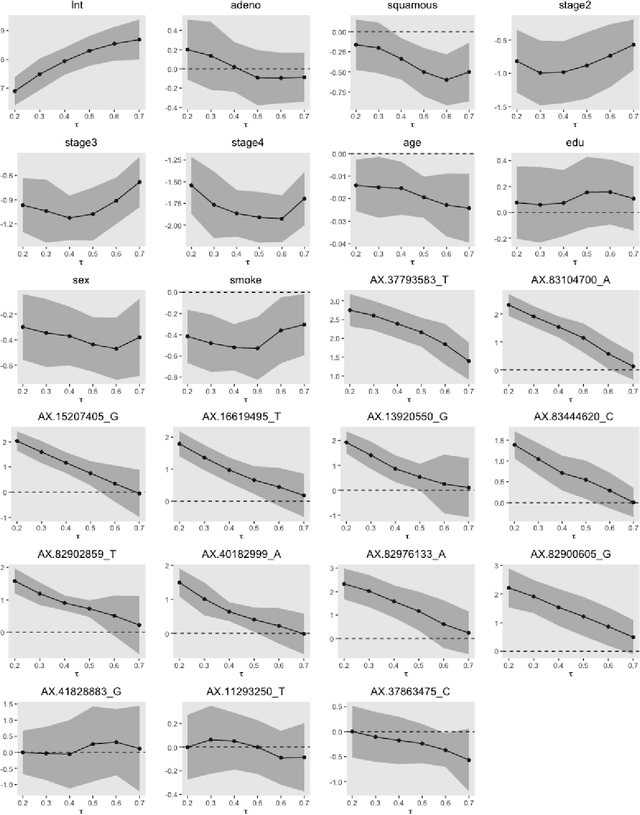
With the availability of high dimensional genetic biomarkers, it is of interest to identify heterogeneous effects of these predictors on patients' survival, along with proper statistical inference. Censored quantile regression has emerged as a powerful tool for detecting heterogeneous effects of covariates on survival outcomes. To our knowledge, there is little work available to draw inference on the effects of high dimensional predictors for censored quantile regression. This paper proposes a novel procedure to draw inference on all predictors within the framework of global censored quantile regression, which investigates covariate-response associations over an interval of quantile levels, instead of a few discrete values. The proposed estimator combines a sequence of low dimensional model estimates that are based on multi-sample splittings and variable selection. We show that, under some regularity conditions, the estimator is consistent and asymptotically follows a Gaussian process indexed by the quantile level. Simulation studies indicate that our procedure can properly quantify the uncertainty of the estimates in high dimensional settings. We apply our method to analyze the heterogeneous effects of SNPs residing in lung cancer pathways on patients' survival, using the Boston Lung Cancer Survival Cohort, a cancer epidemiology study on the molecular mechanism of lung cancer.