 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeZeroDDI: A Zero-Shot Drug-Drug Interaction Event Prediction Method with Semantic Enhanced Learning and Dual-Modal Uniform Alignment
Jul 01, 2024



Drug-drug interactions (DDIs) can result in various pharmacological changes, which can be categorized into different classes known as DDI events (DDIEs). In recent years, previously unobserved/unseen DDIEs have been emerging, posing a new classification task when unseen classes have no labelled instances in the training stage, which is formulated as a zero-shot DDIE prediction (ZS-DDIE) task. However, existing computational methods are not directly applicable to ZS-DDIE, which has two primary challenges: obtaining suitable DDIE representations and handling the class imbalance issue. To overcome these challenges, we propose a novel method named ZeroDDI for the ZS-DDIE task. Specifically, we design a biological semantic enhanced DDIE representation learning module, which emphasizes the key biological semantics and distills discriminative molecular substructure-related semantics for DDIE representation learning. Furthermore, we propose a dual-modal uniform alignment strategy to distribute drug pair representations and DDIE semantic representations uniformly in a unit sphere and align the matched ones, which can mitigate the issue of class imbalance. Extensive experiments showed that ZeroDDI surpasses the baselines and indicate that it is a promising tool for detecting unseen DDIEs. Our code has been released in https://github.com/wzy-Sarah/ZeroDDI.
Heterogeneous Causal Metapath Graph Neural Network for Gene-Microbe-Disease Association Prediction
Jun 27, 2024The recent focus on microbes in human medicine highlights their potential role in the genetic framework of diseases. To decode the complex interactions among genes, microbes, and diseases, computational predictions of gene-microbe-disease (GMD) associations are crucial. Existing methods primarily address gene-disease and microbe-disease associations, but the more intricate triple-wise GMD associations remain less explored. In this paper, we propose a Heterogeneous Causal Metapath Graph Neural Network (HCMGNN) to predict GMD associations. HCMGNN constructs a heterogeneous graph linking genes, microbes, and diseases through their pairwise associations, and utilizes six predefined causal metapaths to extract directed causal subgraphs, which facilitate the multi-view analysis of causal relations among three entity types. Within each subgraph, we employ a causal semantic sharing message passing network for node representation learning, coupled with an attentive fusion method to integrate these representations for predicting GMD associations. Our extensive experiments show that HCMGNN effectively predicts GMD associations and addresses association sparsity issue by enhancing the graph's semantics and structure.
A Multi-Modal Contrastive Diffusion Model for Therapeutic Peptide Generation
Jan 04, 2024Therapeutic peptides represent a unique class of pharmaceutical agents crucial for the treatment of human diseases. Recently, deep generative models have exhibited remarkable potential for generating therapeutic peptides, but they only utilize sequence or structure information alone, which hinders the performance in generation. In this study, we propose a Multi-Modal Contrastive Diffusion model (MMCD), fusing both sequence and structure modalities in a diffusion framework to co-generate novel peptide sequences and structures. Specifically, MMCD constructs the sequence-modal and structure-modal diffusion models, respectively, and devises a multi-modal contrastive learning strategy with intercontrastive and intra-contrastive in each diffusion timestep, aiming to capture the consistency between two modalities and boost model performance. The inter-contrastive aligns sequences and structures of peptides by maximizing the agreement of their embeddings, while the intra-contrastive differentiates therapeutic and non-therapeutic peptides by maximizing the disagreement of their sequence/structure embeddings simultaneously. The extensive experiments demonstrate that MMCD performs better than other state-of-theart deep generative methods in generating therapeutic peptides across various metrics, including antimicrobial/anticancer score, diversity, and peptide-docking.
Predicting microRNA-disease associations from knowledge graph using tensor decomposition with relational constraints
Nov 13, 2019
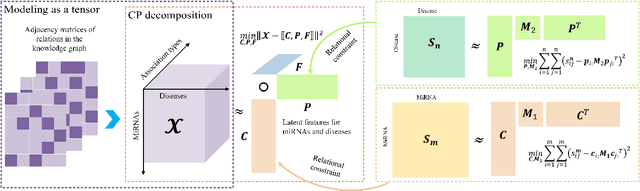
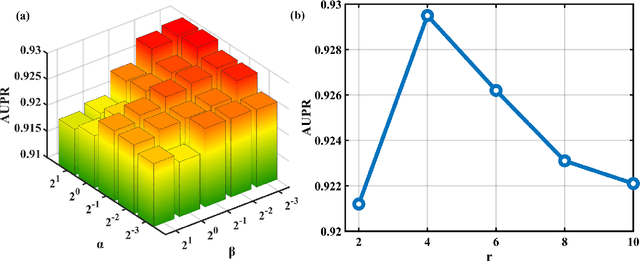
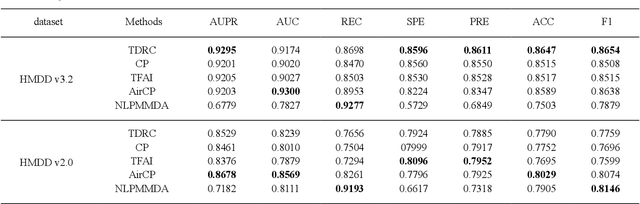
Motivation: MiRNAs are a kind of small non-coding RNAs that are not translated into proteins, and aberrant expression of miRNAs is associated with human diseases. Since miRNAs have different roles in diseases, the miRNA-disease associations are categorized into multiple types according to their roles. Predicting miRNA-disease associations and types is critical to understand the underlying pathogenesis of human diseases from the molecular level. Results: In this paper, we formulate the problem as a link prediction in knowledge graphs. We use biomedical knowledge bases to build a knowledge graph of entities representing miRNAs and disease and multi-relations, and we propose a tensor decomposition-based model named TDRC to predict miRNA-disease associations and their types from the knowledge graph. We have experimentally evaluated our method and compared it to several baseline methods. The results demonstrate that the proposed method has high-accuracy and high-efficiency performances.