 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEPO: Diverse and Realistic Protein Ensemble Generation via Energy Preference Optimization
Nov 13, 2025Accurate exploration of protein conformational ensembles is essential for uncovering function but remains hard because molecular-dynamics (MD) simulations suffer from high computational costs and energy-barrier trapping. This paper presents Energy Preference Optimization (EPO), an online refinement algorithm that turns a pretrained protein ensemble generator into an energy-aware sampler without extra MD trajectories. Specifically, EPO leverages stochastic differential equation sampling to explore the conformational landscape and incorporates a novel energy-ranking mechanism based on list-wise preference optimization. Crucially, EPO introduces a practical upper bound to efficiently approximate the intractable probability of long sampling trajectories in continuous-time generative models, making it easily adaptable to existing pretrained generators. On Tetrapeptides, ATLAS, and Fast-Folding benchmarks, EPO successfully generates diverse and physically realistic ensembles, establishing a new state-of-the-art in nine evaluation metrics. These results demonstrate that energy-only preference signals can efficiently steer generative models toward thermodynamically consistent conformational ensembles, providing an alternative to long MD simulations and widening the applicability of learned potentials in structural biology and drug discovery.
The Latent Road to Atoms: Backmapping Coarse-grained Protein Structures with Latent Diffusion
Oct 17, 2024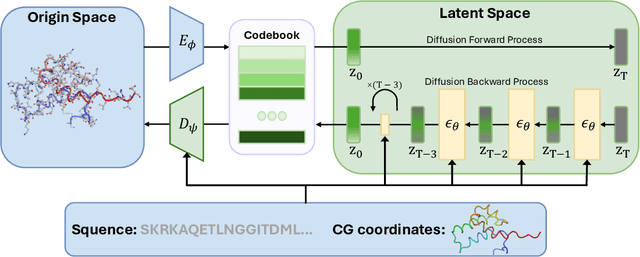
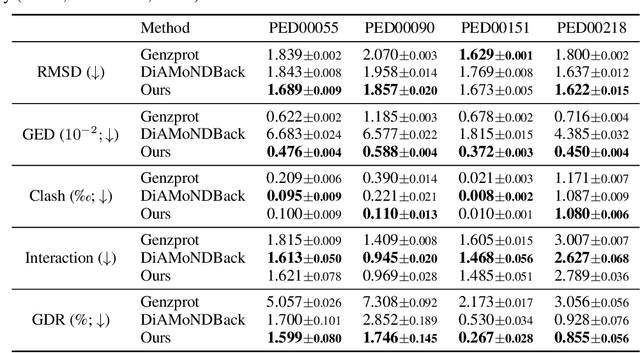
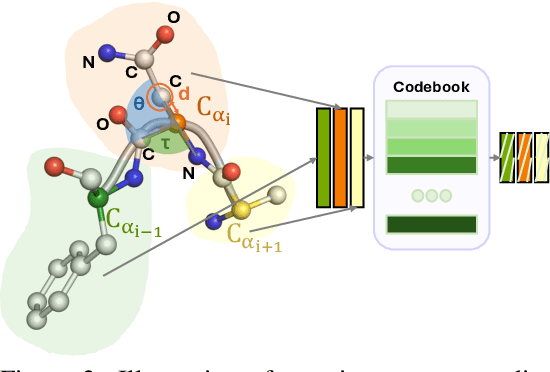
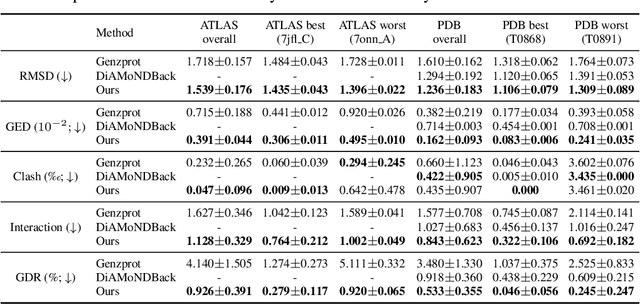
Coarse-grained(CG) molecular dynamics simulations offer computational efficiency for exploring protein conformational ensembles and thermodynamic properties. Though coarse representations enable large-scale simulations across extended temporal and spatial ranges, the sacrifice of atomic-level details limits their utility in tasks such as ligand docking and protein-protein interaction prediction. Backmapping, the process of reconstructing all-atom structures from coarse-grained representations, is crucial for recovering these fine details. While recent machine learning methods have made strides in protein structure generation, challenges persist in reconstructing diverse atomistic conformations that maintain geometric accuracy and chemical validity. In this paper, we present Latent Diffusion Backmapping (LDB), a novel approach leveraging denoising diffusion within latent space to address these challenges. By combining discrete latent encoding with diffusion, LDB bypasses the need for equivariant and internal coordinate manipulation, significantly simplifying the training and sampling processes as well as facilitating better and wider exploration in configuration space. We evaluate LDB's state-of-the-art performance on three distinct protein datasets, demonstrating its ability to efficiently reconstruct structures with high structural accuracy and chemical validity. Moreover, LDB shows exceptional versatility in capturing diverse protein ensembles, highlighting its capability to explore intricate conformational spaces. Our results position LDB as a powerful and scalable approach for backmapping, effectively bridging the gap between CG simulations and atomic-level analyses in computational biology.
Pre-training with Fractional Denoising to Enhance Molecular Property Prediction
Jul 14, 2024Deep learning methods have been considered promising for accelerating molecular screening in drug discovery and material design. Due to the limited availability of labelled data, various self-supervised molecular pre-training methods have been presented. While many existing methods utilize common pre-training tasks in computer vision (CV) and natural language processing (NLP), they often overlook the fundamental physical principles governing molecules. In contrast, applying denoising in pre-training can be interpreted as an equivalent force learning, but the limited noise distribution introduces bias into the molecular distribution. To address this issue, we introduce a molecular pre-training framework called fractional denoising (Frad), which decouples noise design from the constraints imposed by force learning equivalence. In this way, the noise becomes customizable, allowing for incorporating chemical priors to significantly improve molecular distribution modeling. Experiments demonstrate that our framework consistently outperforms existing methods, establishing state-of-the-art results across force prediction, quantum chemical properties, and binding affinity tasks. The refined noise design enhances force accuracy and sampling coverage, which contribute to the creation of physically consistent molecular representations, ultimately leading to superior predictive performance.