 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Latent Road to Atoms: Backmapping Coarse-grained Protein Structures with Latent Diffusion
Paper and Code
Oct 17, 2024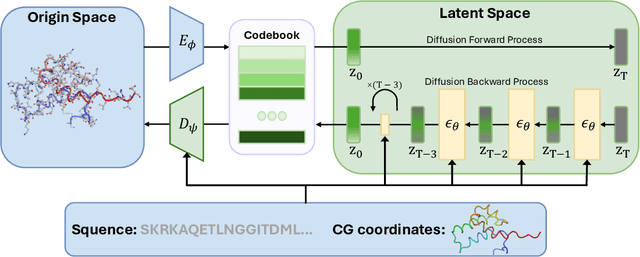
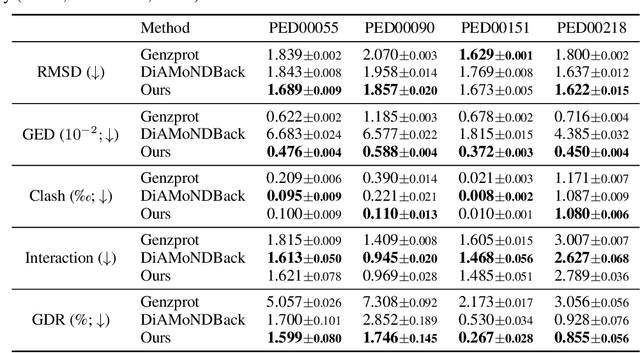
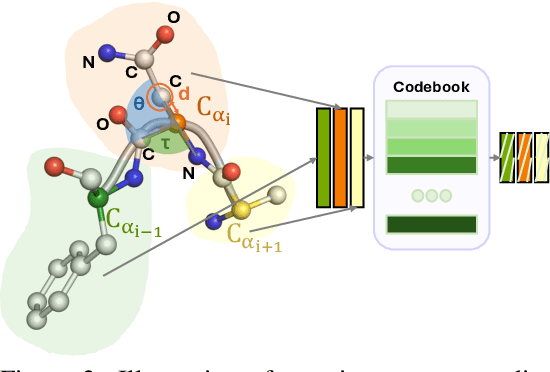
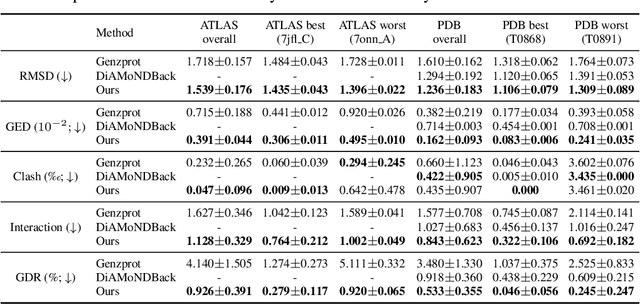
Coarse-grained(CG) molecular dynamics simulations offer computational efficiency for exploring protein conformational ensembles and thermodynamic properties. Though coarse representations enable large-scale simulations across extended temporal and spatial ranges, the sacrifice of atomic-level details limits their utility in tasks such as ligand docking and protein-protein interaction prediction. Backmapping, the process of reconstructing all-atom structures from coarse-grained representations, is crucial for recovering these fine details. While recent machine learning methods have made strides in protein structure generation, challenges persist in reconstructing diverse atomistic conformations that maintain geometric accuracy and chemical validity. In this paper, we present Latent Diffusion Backmapping (LDB), a novel approach leveraging denoising diffusion within latent space to address these challenges. By combining discrete latent encoding with diffusion, LDB bypasses the need for equivariant and internal coordinate manipulation, significantly simplifying the training and sampling processes as well as facilitating better and wider exploration in configuration space. We evaluate LDB's state-of-the-art performance on three distinct protein datasets, demonstrating its ability to efficiently reconstruct structures with high structural accuracy and chemical validity. Moreover, LDB shows exceptional versatility in capturing diverse protein ensembles, highlighting its capability to explore intricate conformational spaces. Our results position LDB as a powerful and scalable approach for backmapping, effectively bridging the gap between CG simulations and atomic-level analyses in computational biology.