 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBoFire: Bayesian Optimization Framework Intended for Real Experiments
Aug 09, 2024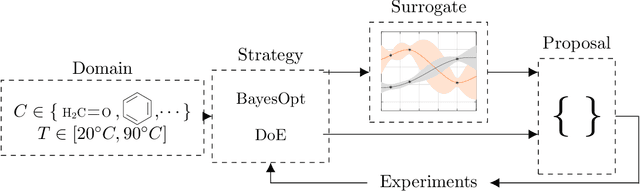
Our open-source Python package BoFire combines Bayesian Optimization (BO) with other design of experiments (DoE) strategies focusing on developing and optimizing new chemistry. Previous BO implementations, for example as they exist in the literature or software, require substantial adaptation for effective real-world deployment in chemical industry. BoFire provides a rich feature-set with extensive configurability and realizes our vision of fast-tracking research contributions into industrial use via maintainable open-source software. Owing to quality-of-life features like JSON-serializability of problem formulations, BoFire enables seamless integration of BO into RESTful APIs, a common architecture component for both self-driving laboratories and human-in-the-loop setups. This paper discusses the differences between BoFire and other BO implementations and outlines ways that BO research needs to be adapted for real-world use in a chemistry setting.
Data-driven modelling of nonlinear dynamics by polytope projections and memory
Dec 13, 2021
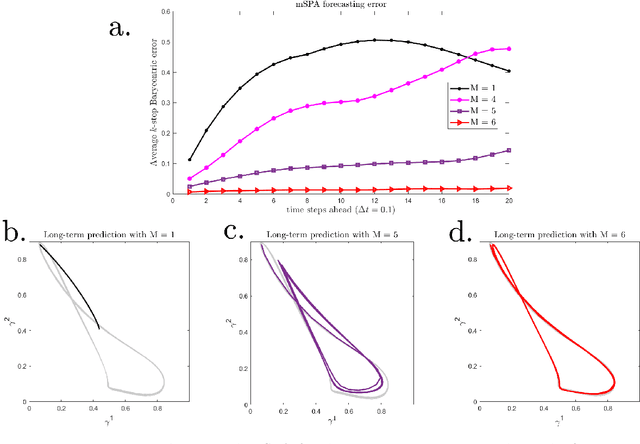


We present a numerical method to model dynamical systems from data. We use the recently introduced method Scalable Probabilistic Approximation (SPA) to project points from a Euclidean space to convex polytopes and represent these projected states of a system in new, lower-dimensional coordinates denoting their position in the polytope. We then introduce a specific nonlinear transformation to construct a model of the dynamics in the polytope and to transform back into the original state space. To overcome the potential loss of information from the projection to a lower-dimensional polytope, we use memory in the sense of the delay-embedding theorem of Takens. By construction, our method produces stable models. We illustrate the capacity of the method to reproduce even chaotic dynamics and attractors with multiple connected components on various examples.
Sparse Proteomics Analysis - A compressed sensing-based approach for feature selection and classification of high-dimensional proteomics mass spectrometry data
Nov 26, 2016


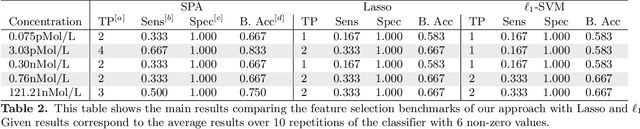
Background: High-throughput proteomics techniques, such as mass spectrometry (MS)-based approaches, produce very high-dimensional data-sets. In a clinical setting one is often interested in how mass spectra differ between patients of different classes, for example spectra from healthy patients vs. spectra from patients having a particular disease. Machine learning algorithms are needed to (a) identify these discriminating features and (b) classify unknown spectra based on this feature set. Since the acquired data is usually noisy, the algorithms should be robust against noise and outliers, while the identified feature set should be as small as possible. Results: We present a new algorithm, Sparse Proteomics Analysis (SPA), based on the theory of compressed sensing that allows us to identify a minimal discriminating set of features from mass spectrometry data-sets. We show (1) how our method performs on artificial and real-world data-sets, (2) that its performance is competitive with standard (and widely used) algorithms for analyzing proteomics data, and (3) that it is robust against random and systematic noise. We further demonstrate the applicability of our algorithm to two previously published clinical data-sets.