 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMaterials-Discovery Workflows Guided by Symbolic Regression: Identifying Acid-Stable Oxides for Electrocatalysis
Dec 08, 2024



The efficiency of active learning (AL) approaches to identify materials with desired properties relies on the knowledge of a few parameters describing the property. However, these parameters are unknown if the property is governed by a high intricacy of many atomistic processes. Here, we develop an AL workflow based on the sure-independence screening and sparsifying operator (SISSO) symbolic-regression approach. SISSO identifies the few, key parameters correlated with a given materials property via analytical expressions, out of many offered primary features. Crucially, we train ensembles of SISSO models in order to quantify mean predictions and their uncertainty, enabling the use of SISSO in AL. By combining bootstrap sampling to obtain training datasets with Monte-Carlo feature dropout, the high prediction errors observed by a single SISSO model are improved. Besides, the feature dropout procedure alleviates the overconfidence issues observed in the widely used bagging approach. We demonstrate the SISSO-guided AL workflow by identifying acid-stable oxides for water splitting using high-quality DFT-HSE06 calculations. From a pool of 1470 materials, 12 acid-stable materials are identified in only 30 AL iterations. The materials property maps provided by SISSO along with the uncertainty estimates reduce the risk of missing promising portions of the materials space that were overlooked in the initial, possibly biased dataset.
Accelerating the Training and Improving the Reliability of Machine-Learned Interatomic Potentials for Strongly Anharmonic Materials through Active Learning
Sep 18, 2024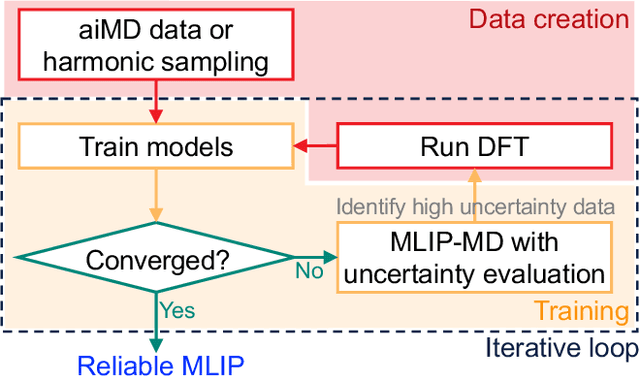

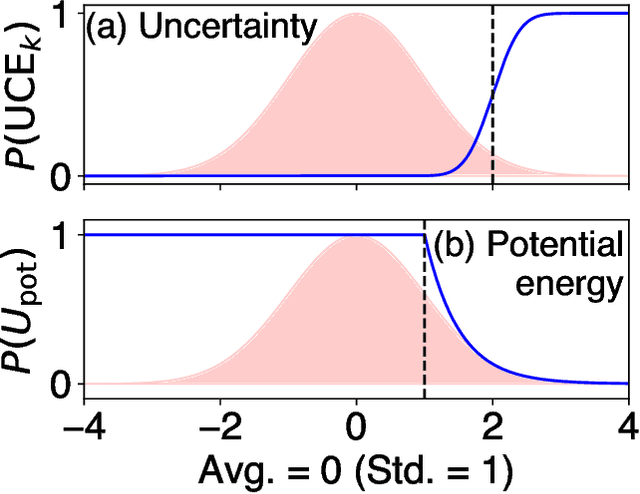
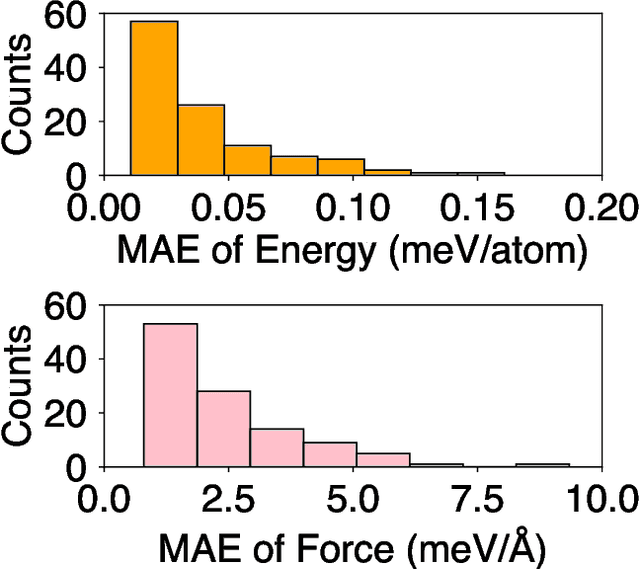
Molecular dynamics (MD) employing machine-learned interatomic potentials (MLIPs) serve as an efficient, urgently needed complement to ab initio molecular dynamics (aiMD). By training these potentials on data generated from ab initio methods, their averaged predictions can exhibit comparable performance to ab initio methods at a fraction of the cost. However, insufficient training sets might lead to an improper description of the dynamics in strongly anharmonic materials, because critical effects might be overlooked in relevant cases, or only incorrectly captured, or hallucinated by the MLIP when they are not actually present. In this work, we show that an active learning scheme that combines MD with MLIPs (MLIP-MD) and uncertainty estimates can avoid such problematic predictions. In short, efficient MLIP-MD is used to explore configuration space quickly, whereby an acquisition function based on uncertainty estimates and on energetic viability is employed to maximize the value of the newly generated data and to focus on the most unfamiliar but reasonably accessible regions of phase space. To verify our methodology, we screen over 112 materials and identify 10 examples experiencing the aforementioned problems. Using CuI and AgGaSe$_2$ as archetypes for these problematic materials, we discuss the physical implications for strongly anharmonic effects and demonstrate how the developed active learning scheme can address these issues.
From Prediction to Action: Critical Role of Performance Estimation for Machine-Learning-Driven Materials Discovery
Dec 07, 2023Materials discovery driven by statistical property models is an iterative decision process, during which an initial data collection is extended with new data proposed by a model-informed acquisition function--with the goal to maximize a certain "reward" over time, such as the maximum property value discovered so far. While the materials science community achieved much progress in developing property models that predict well on average with respect to the training distribution, this form of in-distribution performance measurement is not directly coupled with the discovery reward. This is because an iterative discovery process has a shifting reward distribution that is over-proportionally determined by the model performance for exceptional materials. We demonstrate this problem using the example of bulk modulus maximization among double perovskite oxides. We find that the in-distribution predictive performance suggests random forests as superior to Gaussian process regression, while the results are inverse in terms of the discovery rewards. We argue that the lack of proper performance estimation methods from pre-computed data collections is a fundamental problem for improving data-driven materials discovery, and we propose a novel such estimator that, in contrast to na\"ive reward estimation, successfully predicts Gaussian processes with the "expected improvement" acquisition function as the best out of four options in our demonstrational study for double perovskites. Importantly, it does so without requiring the over thousand ab initio computations that were needed to confirm this prediction.
Extrapolation to complete basis-set limit in density-functional theory by quantile random-forest models
Mar 31, 2023



The numerical precision of density-functional-theory (DFT) calculations depends on a variety of computational parameters, one of the most critical being the basis-set size. The ultimate precision is reached with an infinitely large basis set, i.e., in the limit of a complete basis set (CBS). Our aim in this work is to find a machine-learning model that extrapolates finite basis-size calculations to the CBS limit. We start with a data set of 63 binary solids investigated with two all-electron DFT codes, exciting and FHI-aims, which employ very different types of basis sets. A quantile-random-forest model is used to estimate the total-energy correction with respect to a fully converged calculation as a function of the basis-set size. The random-forest model achieves a symmetric mean absolute percentage error of lower than 25% for both codes and outperforms previous approaches in the literature. Our approach also provides prediction intervals, which quantify the uncertainty of the models' predictions.
Heat flux for semi-local machine-learning potentials
Mar 28, 2023



The Green-Kubo (GK) method is a rigorous framework for heat transport simulations in materials. However, it requires an accurate description of the potential-energy surface and carefully converged statistics. Machine-learning potentials can achieve the accuracy of first-principles simulations while allowing to reach well beyond their simulation time and length scales at a fraction of the cost. In this paper, we explain how to apply the GK approach to the recent class of message-passing machine-learning potentials, which iteratively consider semi-local interactions beyond the initial interaction cutoff. We derive an adapted heat flux formulation that can be implemented using automatic differentiation without compromising computational efficiency. The approach is demonstrated and validated by calculating the thermal conductivity of zirconium dioxide across temperatures.
TCMI: a non-parametric mutual-dependence estimator for multivariate continuous distributions
Jan 30, 2020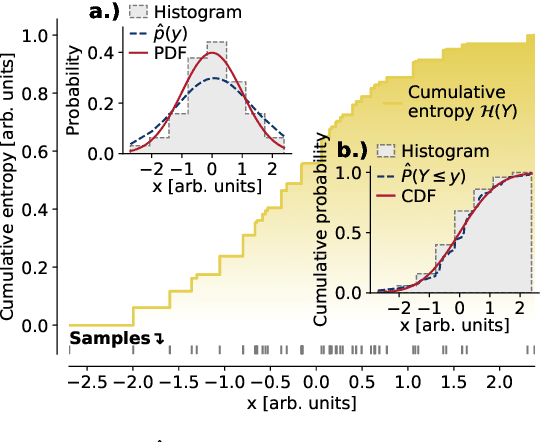

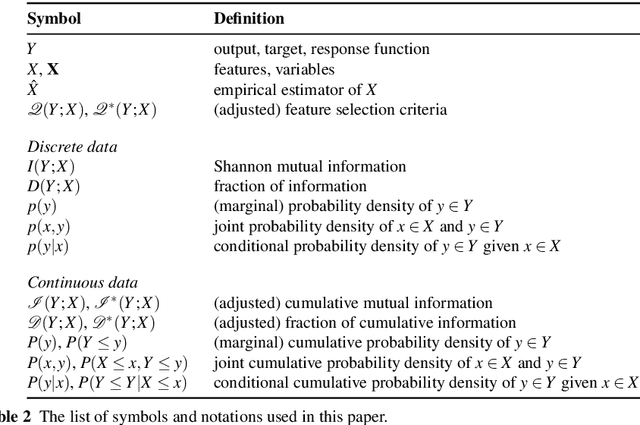
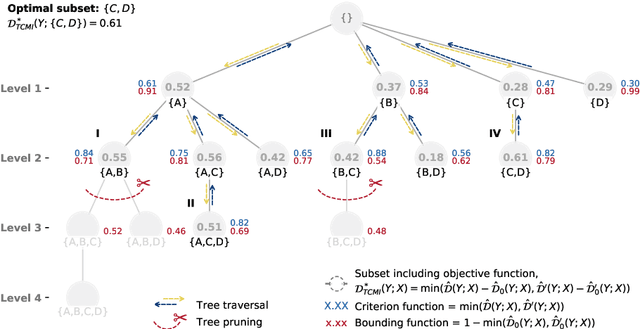
The identification of relevant features, i.e., the driving variables that determine a process or the property of a system, is an essential part of the analysis of data sets whose entries are described by a large number of variables. The preferred measure for quantifying the relevance of nonlinear statistical dependencies is mutual information, which requires as input probability distributions. Probability distributions cannot be reliably sampled and estimated from limited data, especially for real-valued data samples such as lengths or energies. Here, we introduce total cumulative mutual information (TCMI), a measure of the relevance of mutual dependencies based on cumulative probability distributions. TCMI can be estimated directly from sample data and is a non-parametric, robust and deterministic measure that facilitates comparisons and rankings between feature sets with different cardinality. The ranking induced by TCMI allows for feature selection, i.e., the identification of the set of relevant features that are statistical related to the process or the property of a system, while taking into account the number of data samples as well as the cardinality of the feature subsets. We evaluate the performance of our measure with simulated data, compare its performance with similar multivariate dependence measures, and demonstrate the effectiveness of our feature selection method on a set of standard data sets and a typical scenario in materials science.