 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerating the Training and Improving the Reliability of Machine-Learned Interatomic Potentials for Strongly Anharmonic Materials through Active Learning
Paper and Code
Sep 18, 2024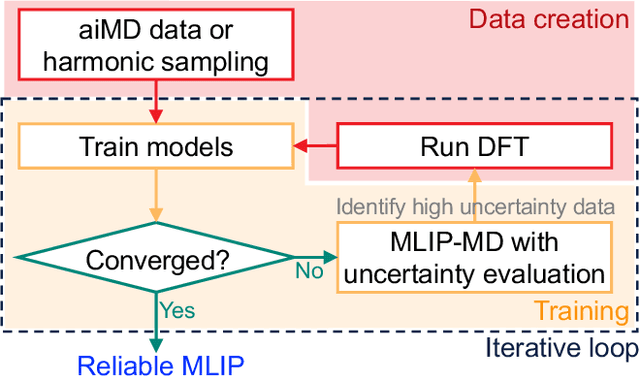

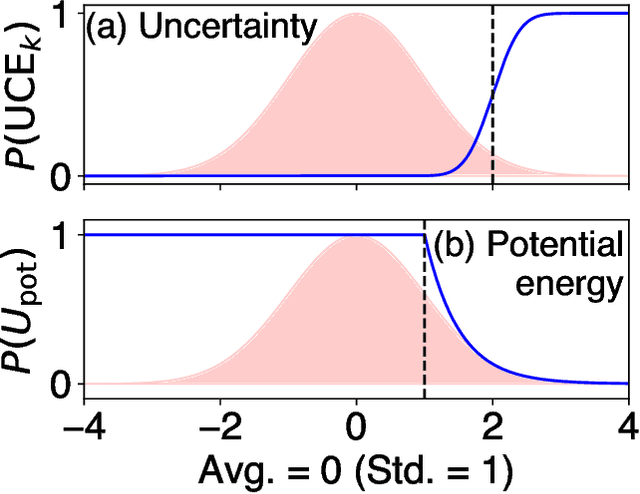
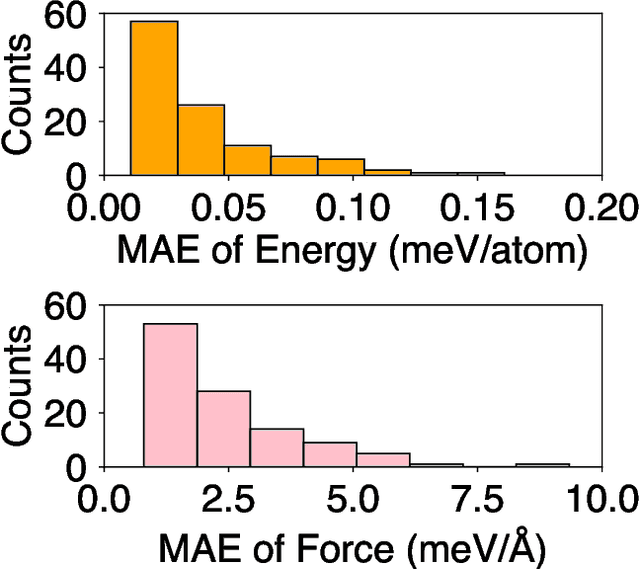
Molecular dynamics (MD) employing machine-learned interatomic potentials (MLIPs) serve as an efficient, urgently needed complement to ab initio molecular dynamics (aiMD). By training these potentials on data generated from ab initio methods, their averaged predictions can exhibit comparable performance to ab initio methods at a fraction of the cost. However, insufficient training sets might lead to an improper description of the dynamics in strongly anharmonic materials, because critical effects might be overlooked in relevant cases, or only incorrectly captured, or hallucinated by the MLIP when they are not actually present. In this work, we show that an active learning scheme that combines MD with MLIPs (MLIP-MD) and uncertainty estimates can avoid such problematic predictions. In short, efficient MLIP-MD is used to explore configuration space quickly, whereby an acquisition function based on uncertainty estimates and on energetic viability is employed to maximize the value of the newly generated data and to focus on the most unfamiliar but reasonably accessible regions of phase space. To verify our methodology, we screen over 112 materials and identify 10 examples experiencing the aforementioned problems. Using CuI and AgGaSe$_2$ as archetypes for these problematic materials, we discuss the physical implications for strongly anharmonic effects and demonstrate how the developed active learning scheme can address these issues.