 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerating Antibiotic Discovery with Large Language Models and Knowledge Graphs
Mar 20, 2025The discovery of novel antibiotics is critical to address the growing antimicrobial resistance (AMR). However, pharmaceutical industries face high costs (over $1 billion), long timelines, and a high failure rate, worsened by the rediscovery of known compounds. We propose an LLM-based pipeline that acts as an alarm system, detecting prior evidence of antibiotic activity to prevent costly rediscoveries. The system integrates organism and chemical literature into a Knowledge Graph (KG), ensuring taxonomic resolution, synonym handling, and multi-level evidence classification. We tested the pipeline on a private list of 73 potential antibiotic-producing organisms, disclosing 12 negative hits for evaluation. The results highlight the effectiveness of the pipeline for evidence reviewing, reducing false negatives, and accelerating decision-making. The KG for negative hits and the user interface for interactive exploration will be made publicly available.
SylloBio-NLI: Evaluating Large Language Models on Biomedical Syllogistic Reasoning
Oct 18, 2024



Syllogistic reasoning is crucial for Natural Language Inference (NLI). This capability is particularly significant in specialized domains such as biomedicine, where it can support automatic evidence interpretation and scientific discovery. This paper presents SylloBio-NLI, a novel framework that leverages external ontologies to systematically instantiate diverse syllogistic arguments for biomedical NLI. We employ SylloBio-NLI to evaluate Large Language Models (LLMs) on identifying valid conclusions and extracting supporting evidence across 28 syllogistic schemes instantiated with human genome pathways. Extensive experiments reveal that biomedical syllogistic reasoning is particularly challenging for zero-shot LLMs, which achieve an average accuracy between 70% on generalized modus ponens and 23% on disjunctive syllogism. At the same time, we found that few-shot prompting can boost the performance of different LLMs, including Gemma (+14%) and LLama-3 (+43%). However, a deeper analysis shows that both techniques exhibit high sensitivity to superficial lexical variations, highlighting a dependency between reliability, models' architecture, and pre-training regime. Overall, our results indicate that, while in-context examples have the potential to elicit syllogistic reasoning in LLMs, existing models are still far from achieving the robustness and consistency required for safe biomedical NLI applications.
An LLM-based Knowledge Synthesis and Scientific Reasoning Framework for Biomedical Discovery
Jun 26, 2024
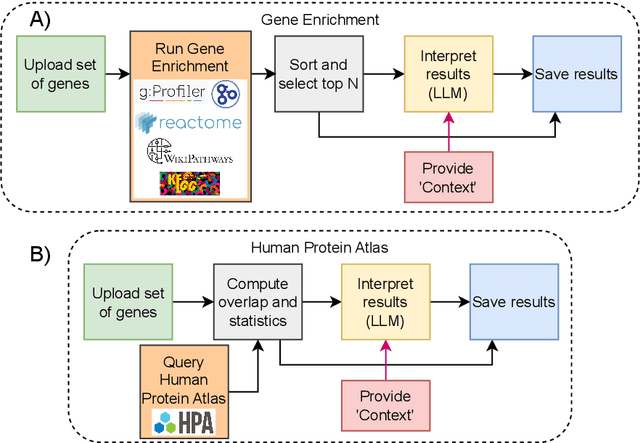
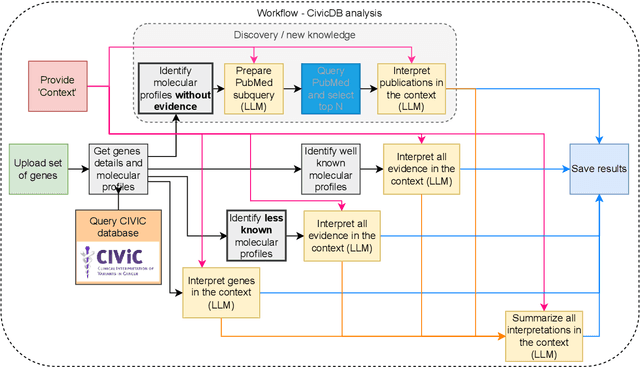
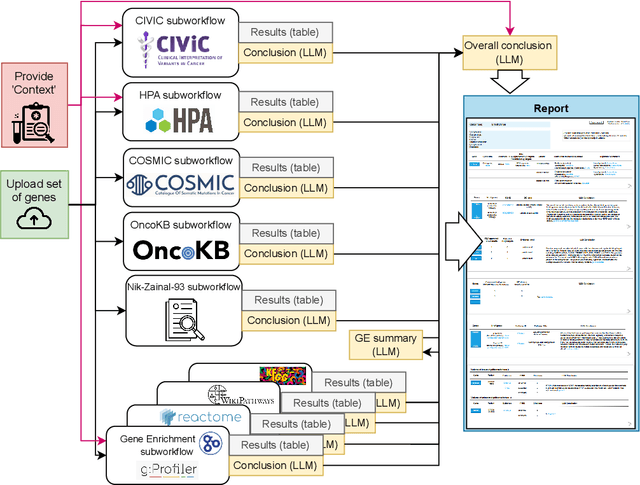
We present BioLunar, developed using the Lunar framework, as a tool for supporting biological analyses, with a particular emphasis on molecular-level evidence enrichment for biomarker discovery in oncology. The platform integrates Large Language Models (LLMs) to facilitate complex scientific reasoning across distributed evidence spaces, enhancing the capability for harmonizing and reasoning over heterogeneous data sources. Demonstrating its utility in cancer research, BioLunar leverages modular design, reusable data access and data analysis components, and a low-code user interface, enabling researchers of all programming levels to construct LLM-enabled scientific workflows. By facilitating automatic scientific discovery and inference from heterogeneous evidence, BioLunar exemplifies the potential of the integration between LLMs, specialised databases and biomedical tools to support expert-level knowledge synthesis and discovery.
Relation Extraction in underexplored biomedical domains: A diversity-optimised sampling and synthetic data generation approach
Nov 10, 2023The sparsity of labelled data is an obstacle to the development of Relation Extraction models and the completion of databases in various biomedical areas. While being of high interest in drug-discovery, the natural-products literature, reporting the identification of potential bioactive compounds from organisms, is a concrete example of such an overlooked topic. To mark the start of this new task, we created the first curated evaluation dataset and extracted literature items from the LOTUS database to build training sets. To this end, we developed a new sampler inspired by diversity metrics in ecology, named Greedy Maximum Entropy sampler, or GME-sampler (https://github.com/idiap/gme-sampler). The strategic optimization of both balance and diversity of the selected items in the evaluation set is important given the resource-intensive nature of manual curation. After quantifying the noise in the training set, in the form of discrepancies between the input abstracts text and the expected output labels, we explored different strategies accordingly. Framing the task as an end-to-end Relation Extraction, we evaluated the performance of standard fine-tuning as a generative task and few-shot learning with open Large Language Models (LLaMA 7B-65B). In addition to their evaluation in few-shot settings, we explore the potential of open Large Language Models (Vicuna-13B) as synthetic data generator and propose a new workflow for this purpose. All evaluated models exhibited substantial improvements when fine-tuned on synthetic abstracts rather than the original noisy data. We provide our best performing (f1-score=59.0) BioGPT-Large model for end-to-end RE of natural-products relationships along with all the generated synthetic data and the evaluation dataset. See more details at https://github.com/idiap/abroad-re.
Large Language Models, scientific knowledge and factuality: A systematic analysis in antibiotic discovery
May 28, 2023Inferring over and extracting information from Large Language Models (LLMs) trained on a large corpus of scientific literature can potentially drive a new era in biomedical research, reducing the barriers for accessing existing medical evidence. This work examines the potential of LLMs for dialoguing with biomedical background knowledge, using the context of antibiotic discovery as an exemplar motivational scenario. The context of biomedical discovery from natural products entails understanding the relational evidence between an organism, an associated chemical and its associated antibiotic properties. We provide a systematic assessment on the ability of LLMs to encode and express these relations, verifying for fluency, prompt-alignment, semantic coherence, factual knowledge and specificity of generated responses. The systematic analysis is applied to nine state-of-the-art models (including ChatGPT and GPT-4) in two prompting-based tasks: chemical compound definition generation and chemical compound-fungus relation determination. Results show that while recent models have improved in fluency, factual accuracy is still low and models are biased towards over-represented entities. The ability of LLMs to serve as biomedical knowledge bases is questioned, and the need for additional systematic evaluation frameworks is highlighted. The best performing GPT-4 produced a factual definition for 70% of chemical compounds and 43.6% factual relations to fungi, whereas the best open source model BioGPT-large 30% of the compounds and 30% of the relations for the best-performing prompt. The results show that while LLMs are currently not fit for purpose to be used as biomedical factual knowledge bases, there is a promising emerging property in the direction of factuality as the models become domain specialised, scale-up in size and level of human feedback.
Biologically-informed deep learning models for cancer: fundamental trends for encoding and interpreting oncology data
Jul 02, 2022



In this paper we provide a structured literature analysis focused on Deep Learning (DL) models used to support inference in cancer biology with a particular emphasis on multi-omics analysis. The work focuses on how existing models address the need for better dialogue with prior knowledge, biological plausibility and interpretability, fundamental properties in the biomedical domain. We discuss the recent evolutionary arch of DL models in the direction of integrating prior biological relational and network knowledge to support better generalisation (e.g. pathways or Protein-Protein-Interaction networks) and interpretability. This represents a fundamental functional shift towards models which can integrate mechanistic and statistical inference aspects. We discuss representational methodologies for the integration of domain prior knowledge in such models. The paper also provides a critical outlook into contemporary methods for explainability and interpretabiltiy. This analysis points in the direction of a convergence between encoding prior knowledge and improved interpretability.
Metareview-informed Explainable Cytokine Storm Detection during CAR-T cell Therapy
Jun 20, 2022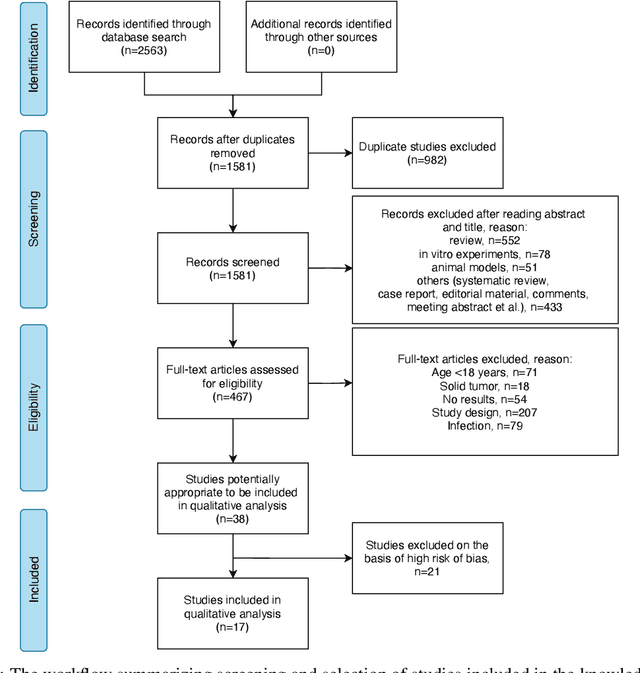
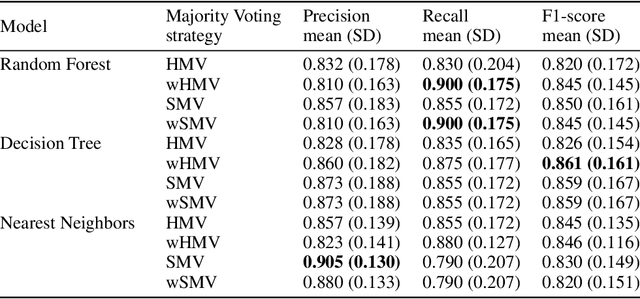
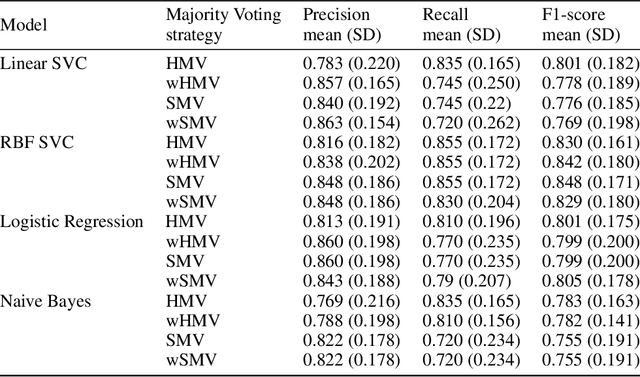
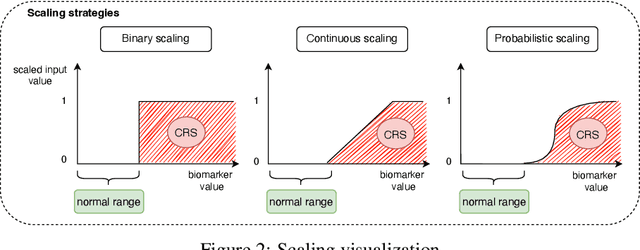
Cytokine release syndrome (CRS), also known as cytokine storm, is one of the most consequential adverse effects of chimeric antigen receptor therapies that have shown promising results in cancer treatment. When emerging, CRS could be identified by the analysis of specific cytokine and chemokine profiles that tend to exhibit similarities across patients. In this paper, we exploit these similarities using machine learning algorithms and set out to pioneer a meta--review informed method for the identification of CRS based on specific cytokine peak concentrations and evidence from previous clinical studies. We argue that such methods could support clinicians in analyzing suspect cytokine profiles by matching them against CRS knowledge from past clinical studies, with the ultimate aim of swift CRS diagnosis. During evaluation with real--world CRS clinical data, we emphasize the potential of our proposed method of producing interpretable results, in addition to being effective in identifying the onset of cytokine storm.
Transformers and the representation of biomedical background knowledge
Feb 04, 2022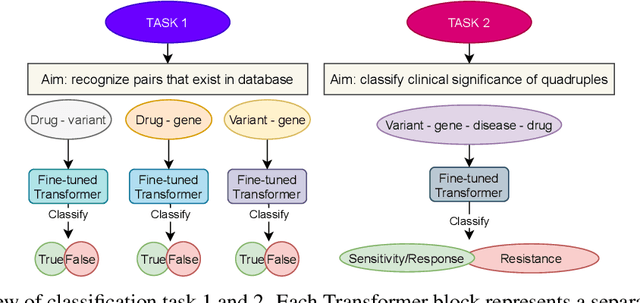

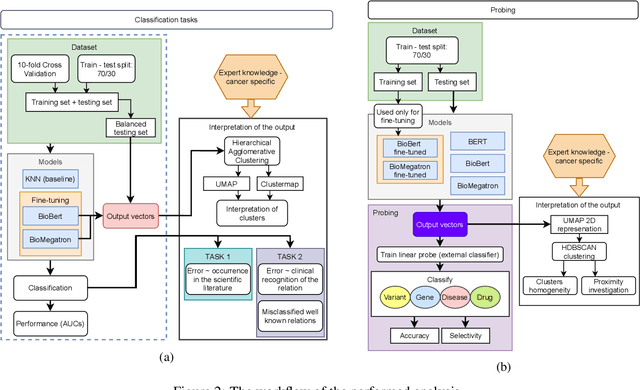
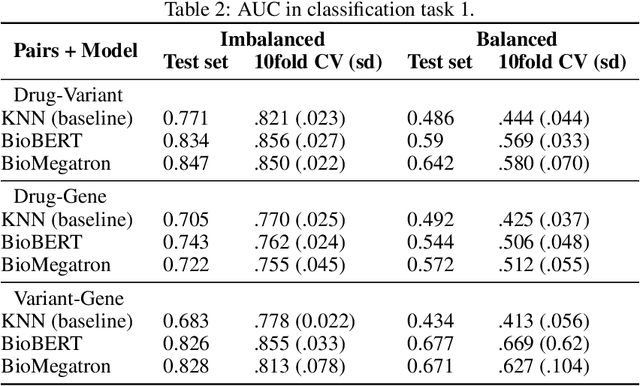
BioBERT and BioMegatron are Transformers models adapted for the biomedical domain based on publicly available biomedical corpora. As such, they have the potential to encode large-scale biological knowledge. We investigate the encoding and representation of biological knowledge in these models, and its potential utility to support inference in cancer precision medicine - namely, the interpretation of the clinical significance of genomic alterations. We compare the performance of different transformer baselines; we use probing to determine the consistency of encodings for distinct entities; and we use clustering methods to compare and contrast the internal properties of the embeddings for genes, variants, drugs and diseases. We show that these models do indeed encode biological knowledge, although some of this is lost in fine-tuning for specific tasks. Finally, we analyse how the models behave with regard to biases and imbalances in the dataset.