 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHeterogeneous Graph based Deep Learning for Biomedical Network Link Prediction
Feb 24, 2021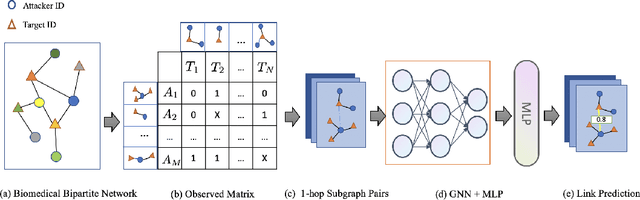
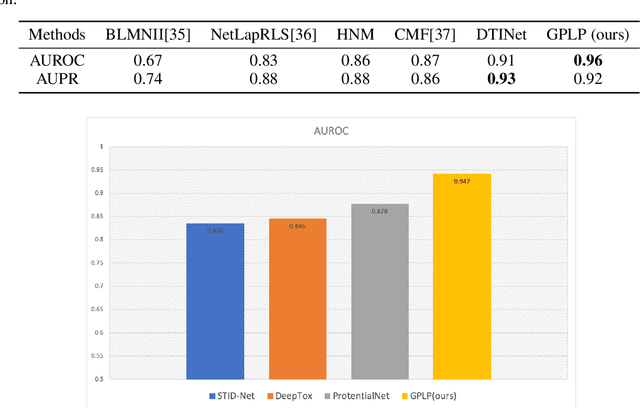
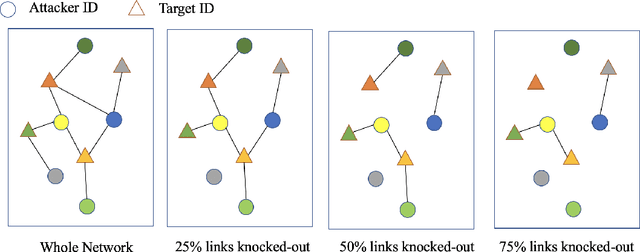
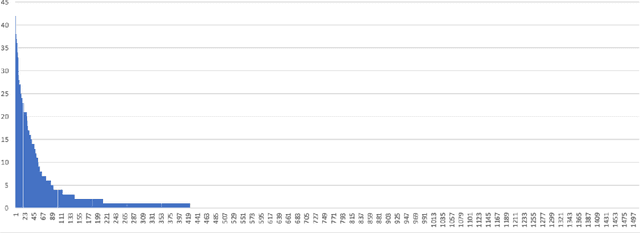
Multi-scale biomedical knowledge networks are expanding with emerging experimental technologies that generates multi-scale biomedical big data. Link prediction is increasingly used especially in bipartite biomedical networks to identify hidden biological interactions and relationshipts between key entities such as compounds, targets, gene and diseases. We propose a Graph Neural Networks (GNN) method, namely Graph Pair based Link Prediction model (GPLP), for predicting biomedical network links simply based on their topological interaction information. In GPLP, 1-hop subgraphs extracted from known network interaction matrix is learnt to predict missing links. To evaluate our method, three heterogeneous biomedical networks were used, i.e. Drug-Target Interaction network (DTI), Compound-Protein Interaction network (CPI) from NIH Tox21, and Compound-Virus Inhibition network (CVI). Our proposed GPLP method significantly outperforms over the state-of-the-art baselines. In addition, different network incompleteness is analysed with our devised protocol, and we also design an effective approach to improve the model robustness towards incomplete networks. Our method demonstrates the potential applications in other biomedical networks.
Sequence-based deep learning antibody design for in silico antibody affinity maturation
Feb 21, 2021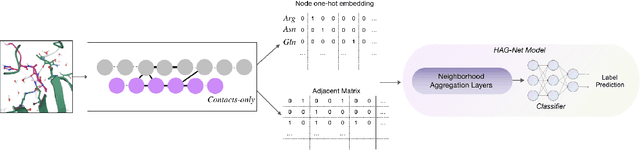
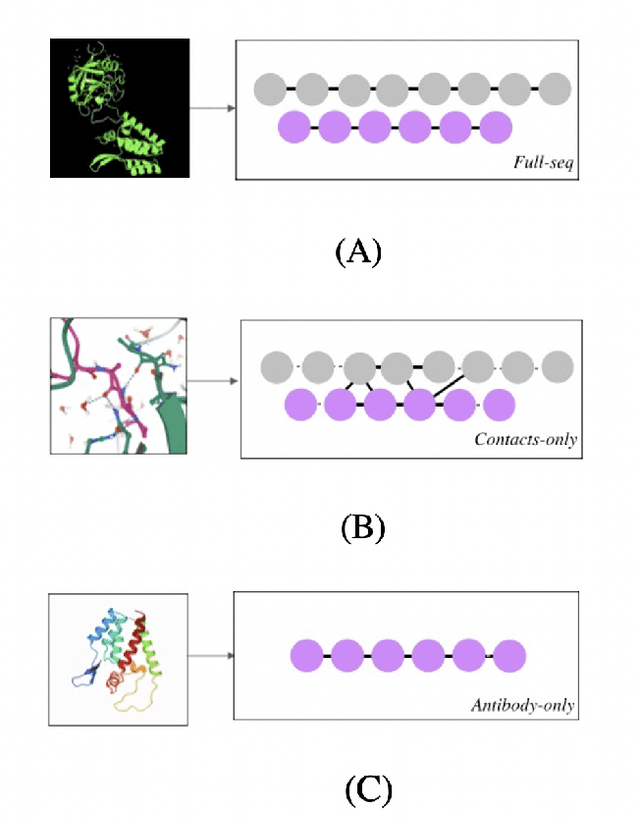
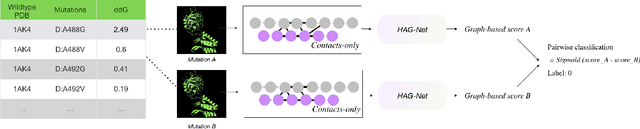
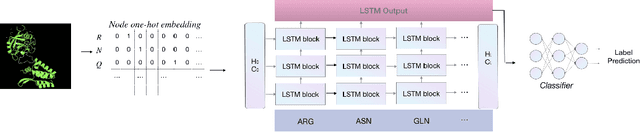
Antibody therapeutics has been extensively studied in drug discovery and development within the past decades. One increasingly popular focus in the antibody discovery pipeline is the optimization step for therapeutic leads. Both traditional methods and in silico approaches aim to generate candidates with high binding affinity against specific target antigens. Traditional in vitro approaches use hybridoma or phage display for candidate selection, and surface plasmon resonance (SPR) for evaluation, while in silico computational approaches aim to reduce the high cost and improve efficiency by incorporating mathematical algorithms and computational processing power in the design process. In the present study, we investigated different graph-based designs for depicting antibody-antigen interactions in terms of antibody affinity prediction using deep learning techniques. While other in silico computations require experimentally determined crystal structures, our study took interest in the capability of sequence-based models for in silico antibody maturation. Our preliminary studies achieved satisfying prediction accuracy on binding affinities comparing to conventional approaches and other deep learning approaches. To further study the antibody-antigen binding specificity, and to simulate the optimization process in real-world scenario, we introduced pairwise prediction strategy. We performed analysis based on both baseline and pairwise prediction results. The resulting prediction and efficiency prove the feasibility and computational efficiency of sequence-based method to be adapted as a scalable industry practice.
Real-time tracking of COVID-19 and coronavirus research updates through text mining
Feb 09, 2021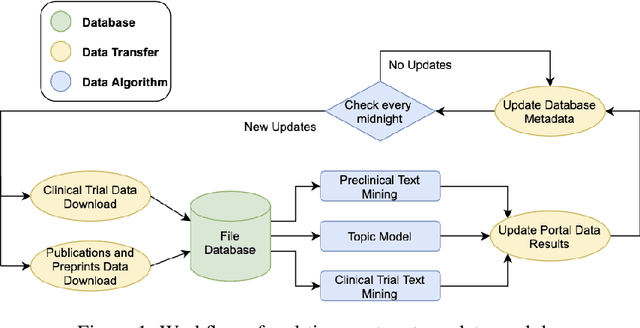
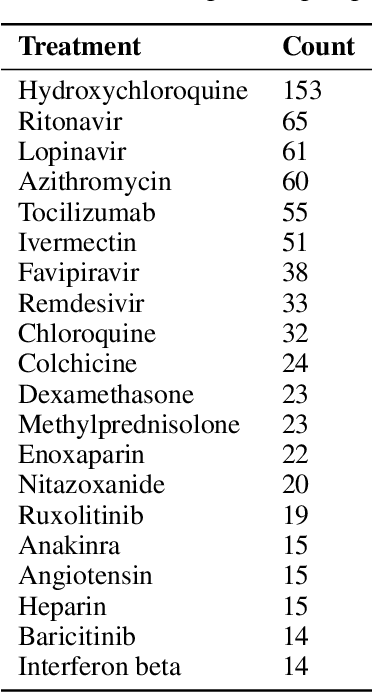
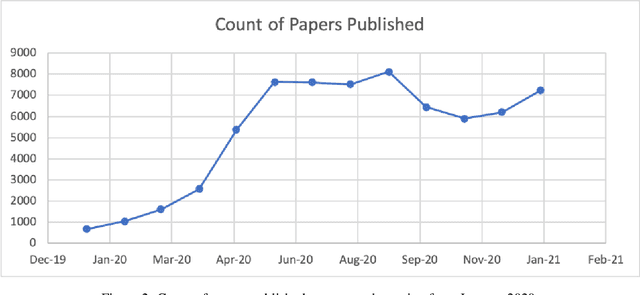
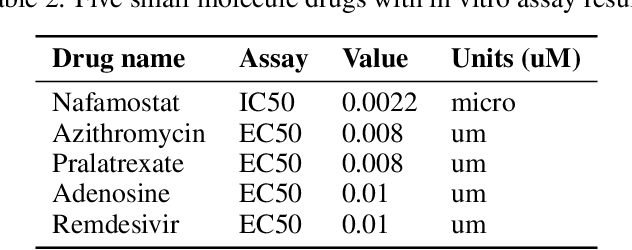
The novel coronavirus (SARS-CoV-2) which causes COVID-19 is an ongoing pandemic. There are ongoing studies with up to hundreds of publications uploaded to databases daily. We are exploring the use-case of artificial intelligence and natural language processing in order to efficiently sort through these publications. We demonstrate that clinical trial information, preclinical studies, and a general topic model can be used as text mining data intelligence tools for scientists all over the world to use as a resource for their own research. To evaluate our method, several metrics are used to measure the information extraction and clustering results. In addition, we demonstrate that our workflow not only have a use-case for COVID-19, but for other disease areas as well. Overall, our system aims to allow scientists to more efficiently research coronavirus. Our automatically updating modules are available on our information portal at https://ghddi-ailab.github.io/Targeting2019-nCoV/ for public viewing.
Enhance Information Propagation for Graph Neural Network by Heterogeneous Aggregations
Feb 08, 2021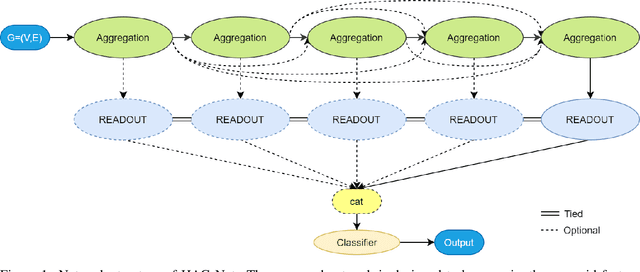

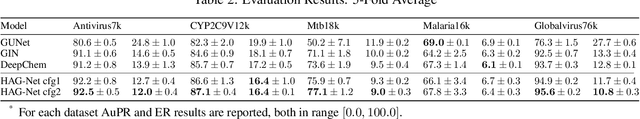
Graph neural networks are emerging as continuation of deep learning success w.r.t. graph data. Tens of different graph neural network variants have been proposed, most following a neighborhood aggregation scheme, where the node features are updated via aggregating features of its neighboring nodes from layer to layer. Though related research surges, the power of GNNs are still not on-par-with their counterpart CNNs in computer vision and RNNs in natural language processing. We rethink this problem from the perspective of information propagation, and propose to enhance information propagation among GNN layers by combining heterogeneous aggregations. We argue that as richer information are propagated from shallow to deep layers, the discriminative capability of features formulated by GNN can benefit from it. As our first attempt in this direction, a new generic GNN layer formulation and upon this a new GNN variant referred as HAG-Net is proposed. We empirically validate the effectiveness of HAG-Net on a number of graph classification benchmarks, and elaborate all the design options and criterions along with.
ParaVS: A Simple, Fast, Efficient and Flexible Graph Neural Network Framework for Structure-Based Virtual Screening
Feb 08, 2021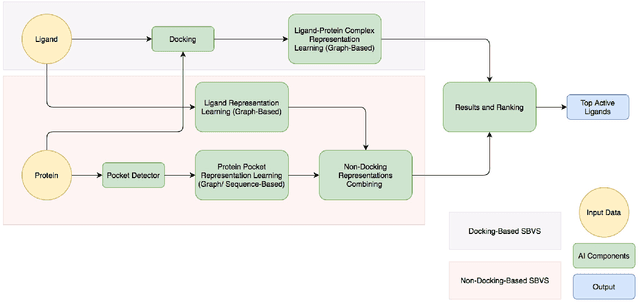

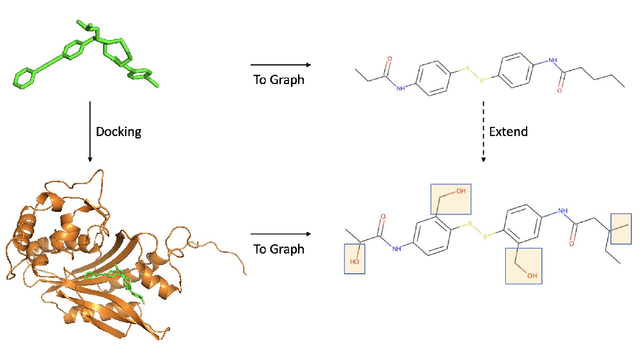

Structure-based virtual screening (SBVS) is a promising in silico technique that integrates computational methods into drug design. An extensively used method in SBVS is molecular docking. However, the docking process can hardly be computationally efficient and accurate simultaneously because classic mechanics scoring function is used to approximate, but hardly reach, the quantum mechanics precision in this method. In order to reduce the computational cost of the protein-ligand scoring process and use data driven approach to boost the scoring function accuracy, we introduce a docking-based SBVS method and, furthermore, a deep learning non-docking-based method that is able to avoid the computational cost of the docking process. Then, we try to integrate these two methods into an easy-to-use framework, ParaVS, that provides both choices for researchers. Graph neural network (GNN) is employed in ParaVS, and we explained how our in-house GNN works and how to model ligands and molecular targets. To verify our approaches, cross validation experiments are done on two datasets, an open dataset Directory of Useful Decoys: Enhanced (DUD.E) and an in-house proprietary dataset without computational generated artificial decoys (NoDecoy). On DUD.E we achieved a state-of-the-art AUC of 0.981 and a state-of-the-art enrichment factor at 2% of 36.2; on NoDecoy we achieved an AUC of 0.974. We further finish inference of an open database, Enamine REAL Database (RDB), that comprises over 1.36 billion molecules in 4050 core-hours using our ParaVS non-docking method (ParaVS-ND). The inference speed of ParaVS-ND is about 3.6e5 molecule / core-hour, while this number of a conventional docking-based method is around 20, which is about 16000 times faster. The experiments indicate that ParaVS is accurate, computationally efficient and can be generalized to different molecular.