 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeParaVS: A Simple, Fast, Efficient and Flexible Graph Neural Network Framework for Structure-Based Virtual Screening
Paper and Code
Feb 08, 2021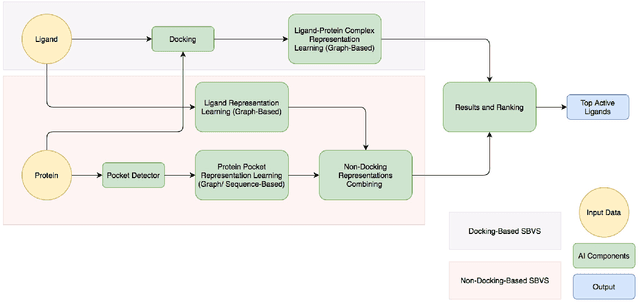

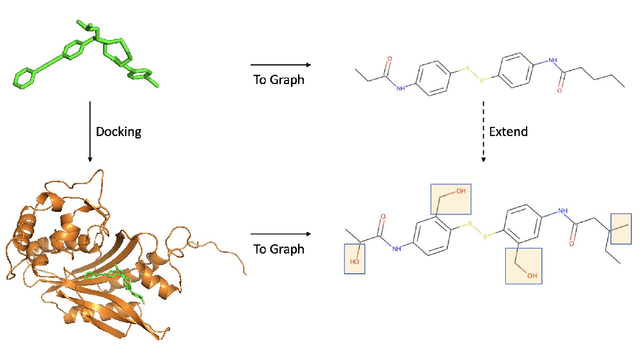

Structure-based virtual screening (SBVS) is a promising in silico technique that integrates computational methods into drug design. An extensively used method in SBVS is molecular docking. However, the docking process can hardly be computationally efficient and accurate simultaneously because classic mechanics scoring function is used to approximate, but hardly reach, the quantum mechanics precision in this method. In order to reduce the computational cost of the protein-ligand scoring process and use data driven approach to boost the scoring function accuracy, we introduce a docking-based SBVS method and, furthermore, a deep learning non-docking-based method that is able to avoid the computational cost of the docking process. Then, we try to integrate these two methods into an easy-to-use framework, ParaVS, that provides both choices for researchers. Graph neural network (GNN) is employed in ParaVS, and we explained how our in-house GNN works and how to model ligands and molecular targets. To verify our approaches, cross validation experiments are done on two datasets, an open dataset Directory of Useful Decoys: Enhanced (DUD.E) and an in-house proprietary dataset without computational generated artificial decoys (NoDecoy). On DUD.E we achieved a state-of-the-art AUC of 0.981 and a state-of-the-art enrichment factor at 2% of 36.2; on NoDecoy we achieved an AUC of 0.974. We further finish inference of an open database, Enamine REAL Database (RDB), that comprises over 1.36 billion molecules in 4050 core-hours using our ParaVS non-docking method (ParaVS-ND). The inference speed of ParaVS-ND is about 3.6e5 molecule / core-hour, while this number of a conventional docking-based method is around 20, which is about 16000 times faster. The experiments indicate that ParaVS is accurate, computationally efficient and can be generalized to different molecular.