 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNeural Networks beyond explainability: Selective inference for sequence motifs
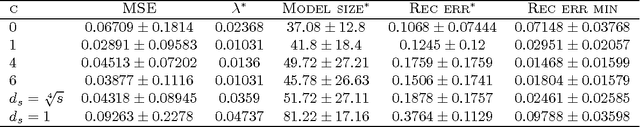
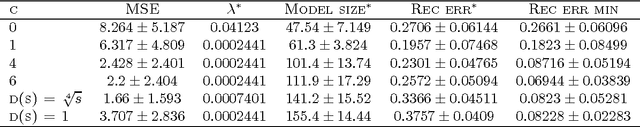
Dec 23, 2022Over the past decade, neural networks have been successful at making predictions from biological sequences, especially in the context of regulatory genomics. As in other fields of deep learning, tools have been devised to extract features such as sequence motifs that can explain the predictions made by a trained network. Here we intend to go beyond explainable machine learning and introduce SEISM, a selective inference procedure to test the association between these extracted features and the predicted phenotype. In particular, we discuss how training a one-layer convolutional network is formally equivalent to selecting motifs maximizing some association score. We adapt existing sampling-based selective inference procedures by quantizing this selection over an infinite set to a large but finite grid. Finally, we show that sampling under a specific choice of parameters is sufficient to characterize the composite null hypothesis typically used for selective inference-a result that goes well beyond our particular framework. We illustrate the behavior of our method in terms of calibration, power and speed and discuss its power/speed trade-off with a simpler data-split strategy. SEISM paves the way to an easier analysis of neural networks used in regulatory genomics, and to more powerful methods for genome wide association studies (GWAS).
Convolutional Kernel Networks for Graph-Structured Data
Mar 11, 2020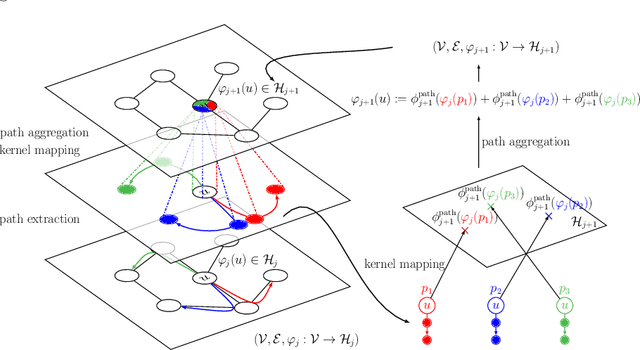
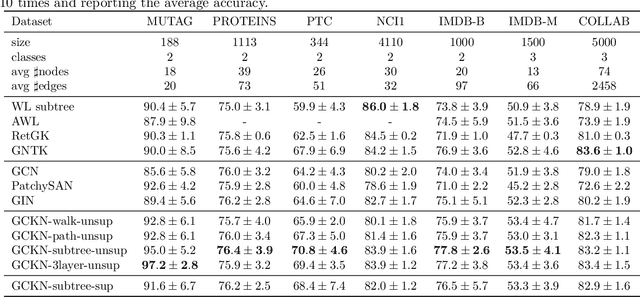
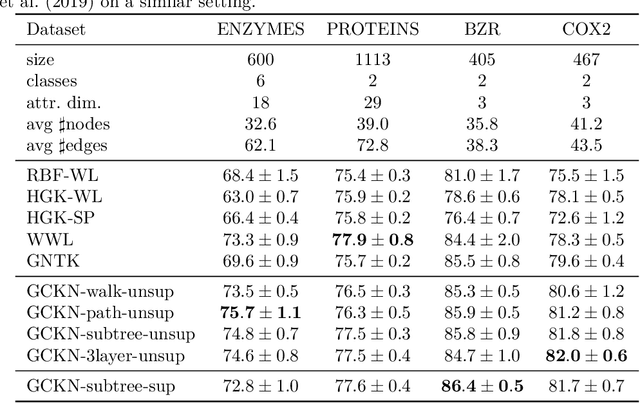
We introduce a family of multilayer graph kernels and establish new links between graph convolutional neural networks and kernel methods. Our approach generalizes convolutional kernel networks to graph-structured data, by representing graphs as a sequence of kernel feature maps, where each node carries information about local graph substructures. On the one hand, the kernel point of view offers an unsupervised, expressive, and easy-to-regularize data representation, which is useful when limited samples are available. On the other hand, our model can also be trained end-to-end on large-scale data, leading to new types of graph convolutional neural networks. We show that our method achieves competitive performance on several graph classification benchmarks, while offering simple model interpretation. Our code is freely available at https://github.com/claying/GCKN.
Recurrent Kernel Networks
Jun 07, 2019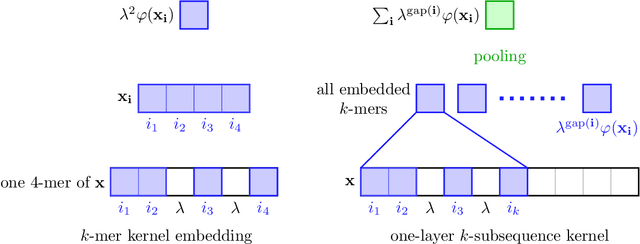
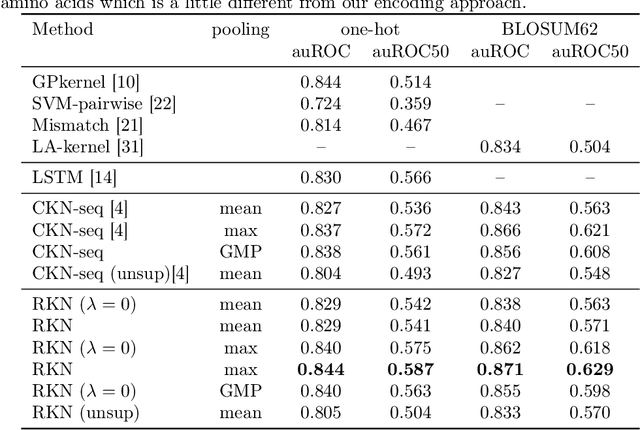
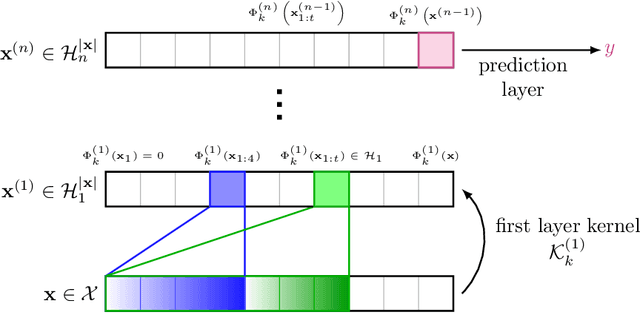

Substring kernels are classical tools for representing biological sequences or text. However, when large amounts of annotated data is available, models that allow end-to-end training such as neural networks are often prefered. Links between recurrent neural networks (RNNs) and substring kernels have recently been drawn, by formally showing that RNNs with specific activation functions were points in a reproducing kernel Hilbert space (RKHS). In this paper, we revisit this link by generalizing convolutional kernel networks---originally related to a relaxation of the mismatch kernel---to model gaps in sequences. It results in a new type of recurrent neural network which can be trained end-to-end with backpropagation, or without supervision by using kernel approximation techniques. We experimentally show that our approach is well suited to biological sequences, where it outperforms existing methods for protein classification tasks.
Group Lasso with Overlaps: the Latent Group Lasso approach
Oct 03, 2011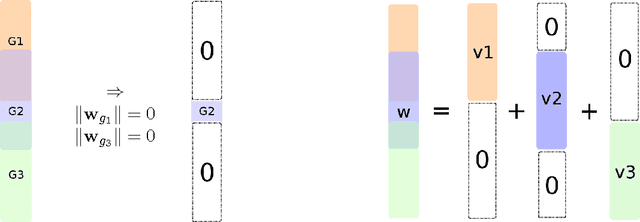
We study a norm for structured sparsity which leads to sparse linear predictors whose supports are unions of prede ned overlapping groups of variables. We call the obtained formulation latent group Lasso, since it is based on applying the usual group Lasso penalty on a set of latent variables. A detailed analysis of the norm and its properties is presented and we characterize conditions under which the set of groups associated with latent variables are correctly identi ed. We motivate and discuss the delicate choice of weights associated to each group, and illustrate this approach on simulated data and on the problem of breast cancer prognosis from gene expression data.
Increasing stability and interpretability of gene expression signatures
Jan 18, 2010

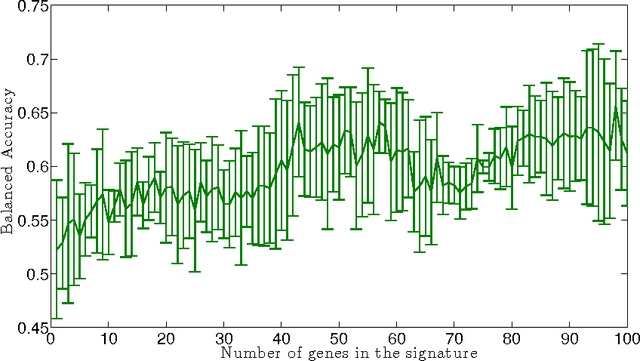

Motivation : Molecular signatures for diagnosis or prognosis estimated from large-scale gene expression data often lack robustness and stability, rendering their biological interpretation challenging. Increasing the signature's interpretability and stability across perturbations of a given dataset and, if possible, across datasets, is urgently needed to ease the discovery of important biological processes and, eventually, new drug targets. Results : We propose a new method to construct signatures with increased stability and easier interpretability. The method uses a gene network as side interpretation and enforces a large connectivity among the genes in the signature, leading to signatures typically made of genes clustered in a few subnetworks. It combines the recently proposed graph Lasso procedure with a stability selection procedure. We evaluate its relevance for the estimation of a prognostic signature in breast cancer, and highlight in particular the increase in interpretability and stability of the signature.
Clustered Multi-Task Learning: A Convex Formulation
Sep 11, 2008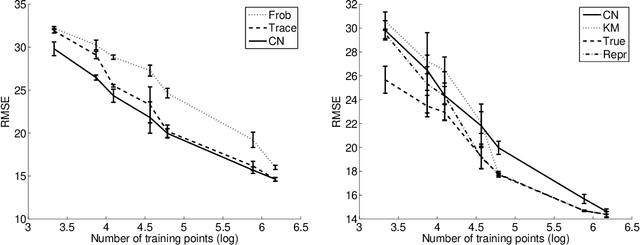
In multi-task learning several related tasks are considered simultaneously, with the hope that by an appropriate sharing of information across tasks, each task may benefit from the others. In the context of learning linear functions for supervised classification or regression, this can be achieved by including a priori information about the weight vectors associated with the tasks, and how they are expected to be related to each other. In this paper, we assume that tasks are clustered into groups, which are unknown beforehand, and that tasks within a group have similar weight vectors. We design a new spectral norm that encodes this a priori assumption, without the prior knowledge of the partition of tasks into groups, resulting in a new convex optimization formulation for multi-task learning. We show in simulations on synthetic examples and on the IEDB MHC-I binding dataset, that our approach outperforms well-known convex methods for multi-task learning, as well as related non convex methods dedicated to the same problem.