 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTIGRESS: Trustful Inference of Gene REgulation using Stability Selection
May 06, 2012



Inferring the structure of gene regulatory networks (GRN) from gene expression data has many applications, from the elucidation of complex biological processes to the identification of potential drug targets. It is however a notoriously difficult problem, for which the many existing methods reach limited accuracy. In this paper, we formulate GRN inference as a sparse regression problem and investigate the performance of a popular feature selection method, least angle regression (LARS) combined with stability selection. We introduce a novel, robust and accurate scoring technique for stability selection, which improves the performance of feature selection with LARS. The resulting method, which we call TIGRESS (Trustful Inference of Gene REgulation using Stability Selection), was ranked among the top methods in the DREAM5 gene network reconstruction challenge. We investigate in depth the influence of the various parameters of the method and show that a fine parameter tuning can lead to significant improvements and state-of-the-art performance for GRN inference. TIGRESS reaches state-of-the-art performance on benchmark data. This study confirms the potential of feature selection techniques for GRN inference. Code and data are available on http://cbio.ensmp.fr/~ahaury. Running TIGRESS online is possible on GenePattern: http://www.broadinstitute.org/cancer/software/genepattern/.
The influence of feature selection methods on accuracy, stability and interpretability of molecular signatures
Jun 23, 2011

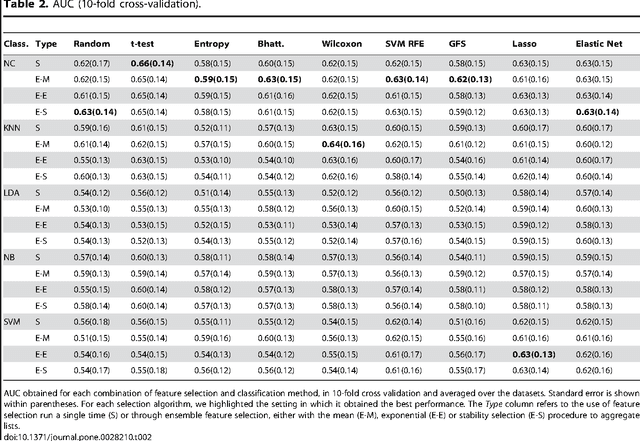

Motivation: Biomarker discovery from high-dimensional data is a crucial problem with enormous applications in biology and medicine. It is also extremely challenging from a statistical viewpoint, but surprisingly few studies have investigated the relative strengths and weaknesses of the plethora of existing feature selection methods. Methods: We compare 32 feature selection methods on 4 public gene expression datasets for breast cancer prognosis, in terms of predictive performance, stability and functional interpretability of the signatures they produce. Results: We observe that the feature selection method has a significant influence on the accuracy, stability and interpretability of signatures. Simple filter methods generally outperform more complex embedded or wrapper methods, and ensemble feature selection has generally no positive effect. Overall a simple Student's t-test seems to provide the best results. Availability: Code and data are publicly available at http://cbio.ensmp.fr/~ahaury/.
Increasing stability and interpretability of gene expression signatures
Jan 18, 2010

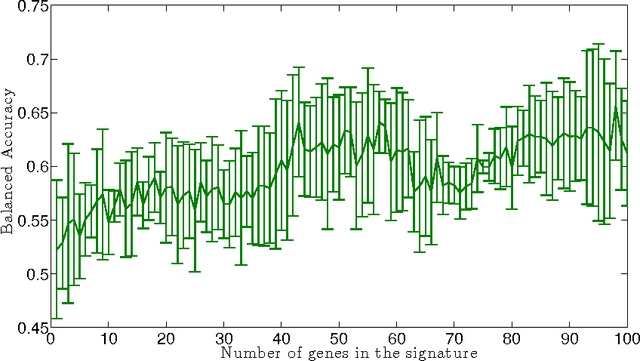

Motivation : Molecular signatures for diagnosis or prognosis estimated from large-scale gene expression data often lack robustness and stability, rendering their biological interpretation challenging. Increasing the signature's interpretability and stability across perturbations of a given dataset and, if possible, across datasets, is urgently needed to ease the discovery of important biological processes and, eventually, new drug targets. Results : We propose a new method to construct signatures with increased stability and easier interpretability. The method uses a gene network as side interpretation and enforces a large connectivity among the genes in the signature, leading to signatures typically made of genes clustered in a few subnetworks. It combines the recently proposed graph Lasso procedure with a stability selection procedure. We evaluate its relevance for the estimation of a prognostic signature in breast cancer, and highlight in particular the increase in interpretability and stability of the signature.