 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDRIVE: Machine Learning to Identify Drivers of Cancer with High-Dimensional Genomic Data & Imputed Labels
May 02, 2021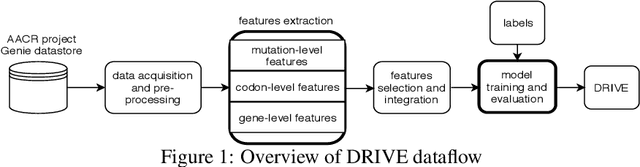
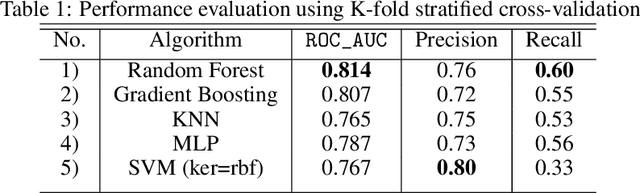
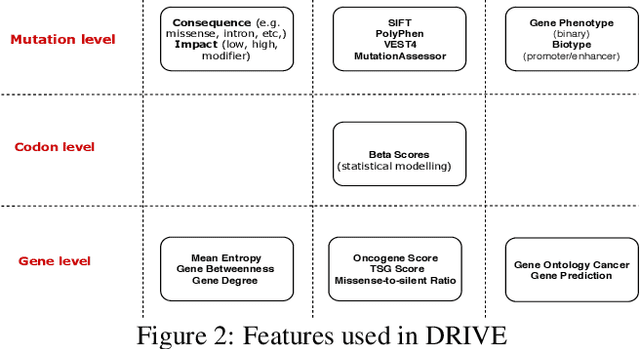

Identifying the mutations that drive cancer growth is key in clinical decision making and precision oncology. As driver mutations confer selective advantage and thus have an increased likelihood of occurrence, frequency-based statistical models are currently favoured. These methods are not suited to rare, low frequency, driver mutations. The alternative approach to address this is through functional-impact scores, however methods using this approach are highly prone to false positives. In this paper, we propose a novel combination method for driver mutation identification, which uses the power of both statistical modelling and functional-impact based methods. Initial results show this approach outperforms the state-of-the-art methods in terms of precision, and provides comparable performance in terms of area under receiver operating characteristic curves (AU-ROC). We believe that data-driven systems based on machine learning, such as these, will become an integral part of precision oncology in the near future.
Deep Bayesian Recurrent Neural Networks for Somatic Variant Calling in Cancer
Dec 06, 2019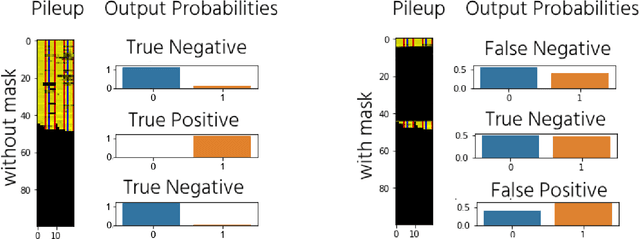
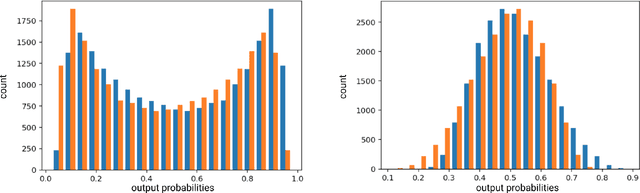
The emerging field of precision oncology relies on the accurate pinpointing of alterations in the molecular profile of a tumor to provide personalized targeted treatments. Current methodologies in the field commonly include the application of next generation sequencing technologies to a tumor sample, followed by the identification of mutations in the DNA known as somatic variants. The differentiation of these variants from sequencing error poses a classic classification problem, which has traditionally been approached with Bayesian statistics, and more recently with supervised machine learning methods such as neural networks. Although these methods provide greater accuracy, classic neural networks lack the ability to indicate the confidence of a variant call. In this paper, we explore the performance of deep Bayesian neural networks on next generation sequencing data, and their ability to give probability estimates for somatic variant calls. In addition to demonstrating similar performance in comparison to standard neural networks, we show that the resultant output probabilities make these better suited to the disparate and highly-variable sequencing data-sets these models are likely to encounter in the real world. We aim to deliver algorithms to oncologists for which model certainty better reflects accuracy, for improved clinical application. By moving away from point estimates to reliable confidence intervals, we expect the resultant clinical and treatment decisions to be more robust and more informed by the underlying reality of the tumor molecular profile.
Safety and Robustness in Decision Making: Deep Bayesian Recurrent Neural Networks for Somatic Variant Calling in Cancer
Dec 04, 2019The genomic profile underlying an individual tumor can be highly informative in the creation of a personalized cancer treatment strategy for a given patient; a practice known as precision oncology. This involves next generation sequencing of a tumor sample and the subsequent identification of genomic aberrations, such as somatic mutations, to provide potential candidates of targeted therapy. The identification of these aberrations from sequencing noise and germline variant background poses a classic classification-style problem. This has been previously broached with many different supervised machine learning methods, including deep-learning neural networks. However, these neural networks have thus far not been tailored to give any indication of confidence in the mutation call, meaning an oncologist could be targeting a mutation with a low probability of being true. To address this, we present here a deep bayesian recurrent neural network for cancer variant calling, which shows no degradation in performance compared to standard neural networks. This approach enables greater flexibility through different priors to avoid overfitting to a single dataset. We will be incorporating this approach into software for oncologists to obtain safe, robust, and statistically confident somatic mutation calls for precision oncology treatment choices.
Effective Sub-clonal Cancer Representation to Predict Tumor Evolution
Nov 28, 2019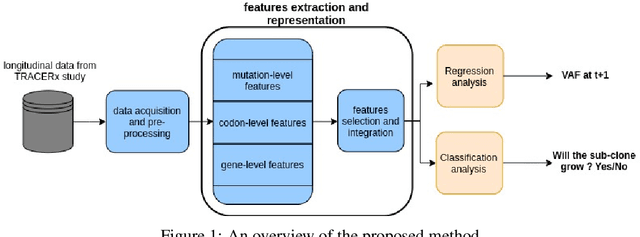
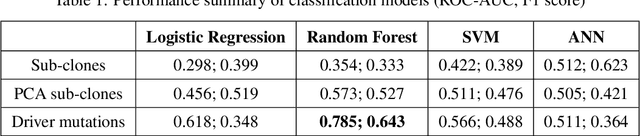
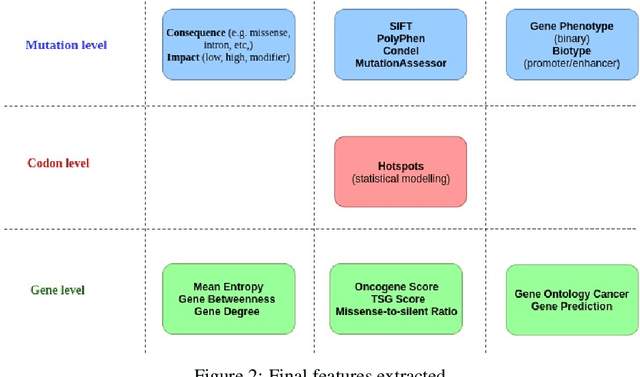
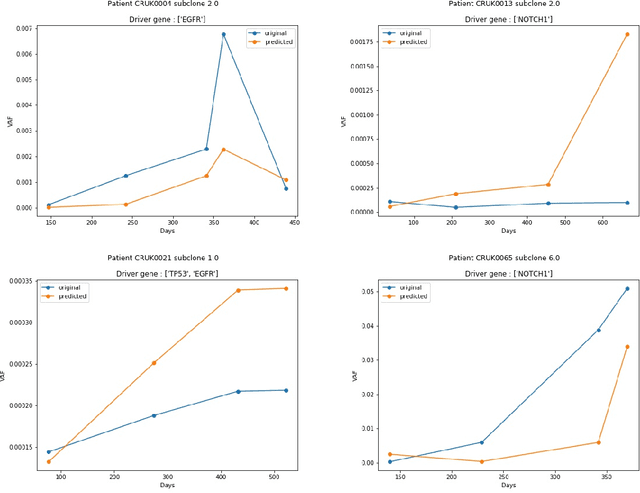
The majority of cancer treatments end in failure due to Intra-Tumor Heterogeneity (ITH). ITH in cancer is represented by clonal evolution where different sub-clones compete with each other for resources under conditions of Darwinian natural selection. Predicting the growth of these sub-clones within a tumour is among the key challenges of modern cancer research. Predicting tumor behavior enables the creation of risk profiles for patients and the optimisation of their treatment by therapeutically targeting sub-clones more likely to grow. Current research efforts in this space are focused on mathematical modelling of population genetics to quantify the selective advantage of sub-clones, thus enabling predictions of which sub-clones are more likely to grow. These tumor evolution models are based on assumptions which are not valid for real-world tumor micro-environment. Furthermore, these models are often fit on a single instance of a tumor, and hence prediction models cannot be validated. This paper presents an alternative approach for predicting cancer evolution using a data-driven machine learning method. Our proposed method is based on the intuition that if we can capture the true characteristics of sub-clones within a tumor and represent it in the form of features, a sophisticated machine learning algorithm can be trained to predict its behavior. The work presented here provides a novel approach to predicting cancer evolution, utilizing a data-driver approach. We strongly believe that the accumulation of data from microbiologists, oncologists and machine learning researchers could be used to encapsulate the true essence of tumor sub-clones, and can play a vital role in selecting the best cancer treatments for patients.
Flatsomatic: A Method for Compression of Somatic Mutation Profiles in Cancer
Nov 27, 2019



In this study, we present Flatsomatic - a Variational Auto Encoder (VAE) optimized to compress somatic mutations that allow for unbiased data compression whilst maintaining the signal. We compared two different neural network architectures for the VAE: Multilayer Perceptron (MLP) and bidirectional LSTM. The somatic profiles we used to train our models consisted of 8,062 Pan-Cancer patients from The Cancer Genome Atlas and 989 cell lines from the COSMIC cell line project. The profiles for each patient were represented by the genomic loci where somatic mutations occurred and, to reduce sparsity, the locations with a frequency <5 were removed. We enhanced the VAE performance by changing its evidence lower bound, and devised an F1-score based loss showing that it helps the VAE learn better than with binary cross-entropy. We also employed beta-VAE to weight the variational regularisation term in the loss function and showed the best performance through a preliminary function to increase the weight of the regularisation term with each epoch. We assessed the reconstruction ability of the VAE using the micro F1-score metric and showed that our best performing model was a 2-layer deep MLP VAE. Our analysis also showed that the size of the latent space did not have a significant effect on the VAE learning ability. We compared the Flatsomatic embeddings created to a lower dimension version of the data from principal component analysis, showing superior performance of Flatsomatic, and performed K-means clustering on both datasets to draw comparisons to known cancer types of each profile. Finally, we present results that confirm that the Flatsomatic representations of 64 dimensions maintain the same predictive power as the original 8,298 dimensions vector, through prediction of drug response.
Learning Embeddings from Cancer Mutation Sets for Classification Tasks
Nov 20, 2019



Analysis of somatic mutation profiles from cancer patients is essential in the development of cancer research. However, the low frequency of most mutations and the varying rates of mutations across patients makes the data extremely challenging to statistically analyze as well as difficult to use in classification problems, for clustering, visualization or for learning useful information. Thus, the creation of low dimensional representations of somatic mutation profiles that hold useful information about the DNA of cancer cells will facilitate the use of such data in applications that will progress precision medicine. In this paper, we talk about the open problem of learning from somatic mutations, and present Flatsomatic: a solution that utilizes variational autoencoders (VAEs) to create latent representations of somatic profiles. The work done in this paper shows great potential for this method, with the VAE embeddings performing better than PCA for a clustering task, and performing equally well to the raw high dimensional data for a classification task. We believe the methods presented herein can be of great value in future research and in bringing data-driven models into precision oncology.
Interlacing Personal and Reference Genomes for Machine Learning Disease-Variant Detection
Nov 26, 2018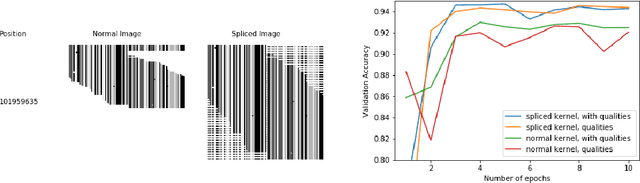
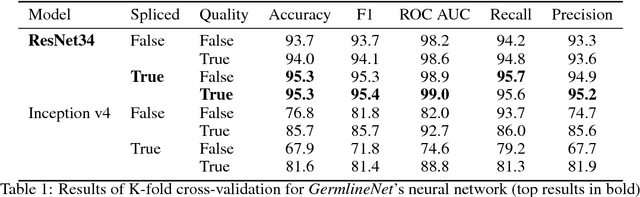
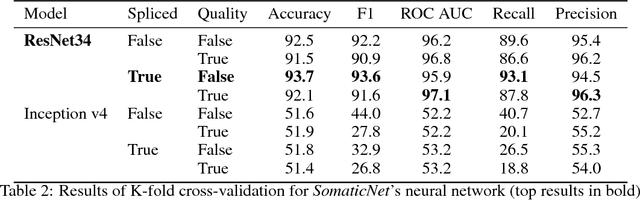
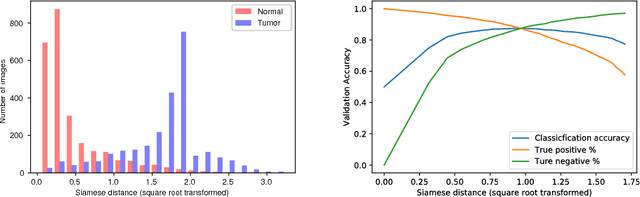
DNA sequencing to identify genetic variants is becoming increasingly valuable in clinical settings. Assessment of variants in such sequencing data is commonly implemented through Bayesian heuristic algorithms. Machine learning has shown great promise in improving on these variant calls, but the input for these is still a standardized "pile-up" image, which is not always best suited. In this paper, we present a novel method for generating images from DNA sequencing data, which interlaces the human reference genome with personalized sequencing output, to maximize usage of sequencing reads and improve machine learning algorithm performance. We demonstrate the success of this in improving standard germline variant calling. We also furthered this approach to include somatic variant calling across tumor/normal data with Siamese networks. These approaches can be used in machine learning applications on sequencing data with the hope of improving clinical outcomes, and are freely available for noncommercial use at www.ccg.ai.
A Framework for Implementing Machine Learning on Omics Data
Nov 26, 2018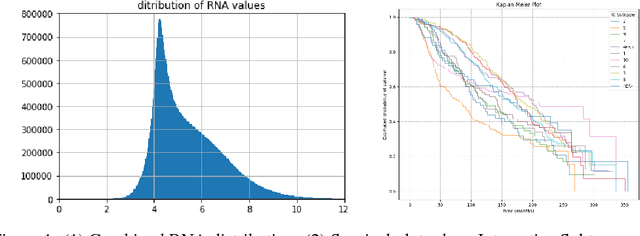
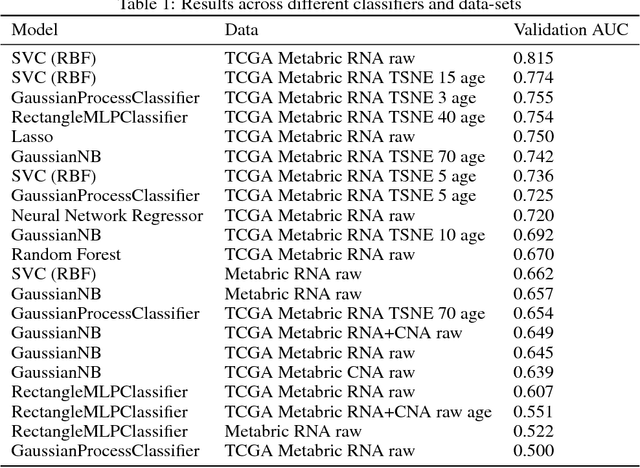
The potential benefits of applying machine learning methods to -omics data are becoming increasingly apparent, especially in clinical settings. However, the unique characteristics of these data are not always well suited to machine learning techniques. These data are often generated across different technologies in different labs, and frequently with high dimensionality. In this paper we present a framework for combining -omics data sets, and for handling high dimensional data, making -omics research more accessible to machine learning applications. We demonstrate the success of this framework through integration and analysis of multi-analyte data for a set of 3,533 breast cancers. We then use this data-set to predict breast cancer patient survival for individuals at risk of an impending event, with higher accuracy and lower variance than methods trained on individual data-sets. We hope that our pipelines for data-set generation and transformation will open up -omics data to machine learning researchers. We have made these freely available for noncommercial use at www.ccg.ai.