 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFloydNet: A Learning Paradigm for Global Relational Reasoning
Jan 27, 2026Developing models capable of complex, multi-step reasoning is a central goal in artificial intelligence. While representing problems as graphs is a powerful approach, Graph Neural Networks (GNNs) are fundamentally constrained by their message-passing mechanism, which imposes a local bottleneck that limits global, holistic reasoning. We argue that dynamic programming (DP), which solves problems by iteratively refining a global state, offers a more powerful and suitable learning paradigm. We introduce FloydNet, a new architecture that embodies this principle. In contrast to local message passing, FloydNet maintains a global, all-pairs relationship tensor and learns a generalized DP operator to progressively refine it. This enables the model to develop a task-specific relational calculus, providing a principled framework for capturing long-range dependencies. Theoretically, we prove that FloydNet achieves 3-WL (2-FWL) expressive power, and its generalized form aligns with the k-FWL hierarchy. FloydNet demonstrates state-of-the-art performance across challenging domains: it achieves near-perfect scores (often >99\%) on the CLRS-30 algorithmic benchmark, finds exact optimal solutions for the general Traveling Salesman Problem (TSP) at rates significantly exceeding strong heuristics, and empirically matches the 3-WL test on the BREC benchmark. Our results establish this learned, DP-style refinement as a powerful and practical alternative to message passing for high-level graph reasoning.
SGNet: Folding Symmetrical Protein Complex with Deep Learning
Mar 07, 2024Deep learning has made significant progress in protein structure prediction, advancing the development of computational biology. However, despite the high accuracy achieved in predicting single-chain structures, a significant number of large homo-oligomeric assemblies exhibit internal symmetry, posing a major challenge in structure determination. The performances of existing deep learning methods are limited since the symmetrical protein assembly usually has a long sequence, making structural computation infeasible. In addition, multiple identical subunits in symmetrical protein complex cause the issue of supervision ambiguity in label assignment, requiring a consistent structure modeling for the training. To tackle these problems, we propose a protein folding framework called SGNet to model protein-protein interactions in symmetrical assemblies. SGNet conducts feature extraction on a single subunit and generates the whole assembly using our proposed symmetry module, which largely mitigates computational problems caused by sequence length. Thanks to the elaborate design of modeling symmetry consistently, we can model all global symmetry types in quaternary protein structure prediction. Extensive experimental results on a benchmark of symmetrical protein complexes further demonstrate the effectiveness of our method.
CMU LiveMedQA at TREC 2017 LiveQA: A Consumer Health Question Answering System
Nov 15, 2017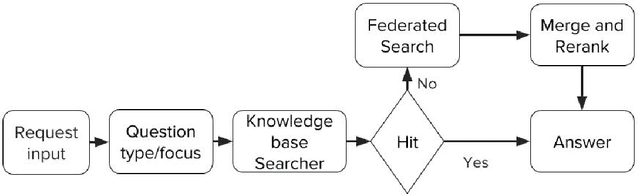



In this paper, we present LiveMedQA, a question answering system that is optimized for consumer health question. On top of the general QA system pipeline, we introduce several new features that aim to exploit domain-specific knowledge and entity structures for better performance. This includes a question type/focus analyzer based on deep text classification model, a tree-based knowledge graph for answer generation and a complementary structure-aware searcher for answer retrieval. LiveMedQA system is evaluated in the TREC 2017 LiveQA medical subtask, where it received an average score of 0.356 on a 3 point scale. Evaluation results revealed 3 substantial drawbacks in current LiveMedQA system, based on which we provide a detailed discussion and propose a few solutions that constitute the main focus of our subsequent work.
Asynchronous Stochastic Proximal Optimization Algorithms with Variance Reduction
Sep 27, 2016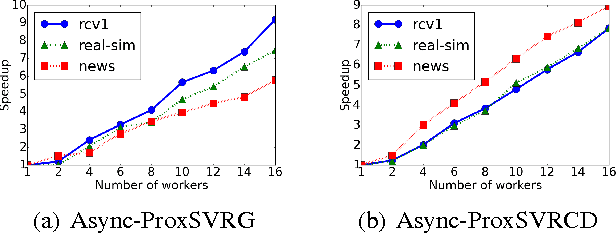

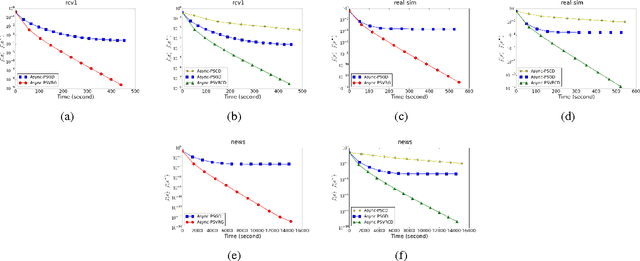
Regularized empirical risk minimization (R-ERM) is an important branch of machine learning, since it constrains the capacity of the hypothesis space and guarantees the generalization ability of the learning algorithm. Two classic proximal optimization algorithms, i.e., proximal stochastic gradient descent (ProxSGD) and proximal stochastic coordinate descent (ProxSCD) have been widely used to solve the R-ERM problem. Recently, variance reduction technique was proposed to improve ProxSGD and ProxSCD, and the corresponding ProxSVRG and ProxSVRCD have better convergence rate. These proximal algorithms with variance reduction technique have also achieved great success in applications at small and moderate scales. However, in order to solve large-scale R-ERM problems and make more practical impacts, the parallel version of these algorithms are sorely needed. In this paper, we propose asynchronous ProxSVRG (Async-ProxSVRG) and asynchronous ProxSVRCD (Async-ProxSVRCD) algorithms, and prove that Async-ProxSVRG can achieve near linear speedup when the training data is sparse, while Async-ProxSVRCD can achieve near linear speedup regardless of the sparse condition, as long as the number of block partitions are appropriately set. We have conducted experiments on a regularized logistic regression task. The results verified our theoretical findings and demonstrated the practical efficiency of the asynchronous stochastic proximal algorithms with variance reduction.