 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlow-Matching Based Refiner for Molecular Conformer Generation
Oct 06, 2025Low-energy molecular conformers generation (MCG) is a foundational yet challenging problem in drug discovery. Denoising-based methods include diffusion and flow-matching methods that learn mappings from a simple base distribution to the molecular conformer distribution. However, these approaches often suffer from error accumulation during sampling, especially in the low SNR steps, which are hard to train. To address these challenges, we propose a flow-matching refiner for the MCG task. The proposed method initializes sampling from mixed-quality outputs produced by upstream denoising models and reschedules the noise scale to bypass the low-SNR phase, thereby improving sample quality. On the GEOM-QM9 and GEOM-Drugs benchmark datasets, the generator-refiner pipeline improves quality with fewer total denoising steps while preserving diversity.
RESTRAIN: From Spurious Votes to Signals -- Self-Driven RL with Self-Penalization
Oct 02, 2025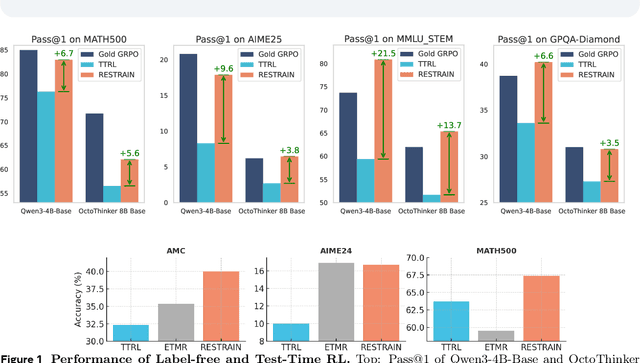
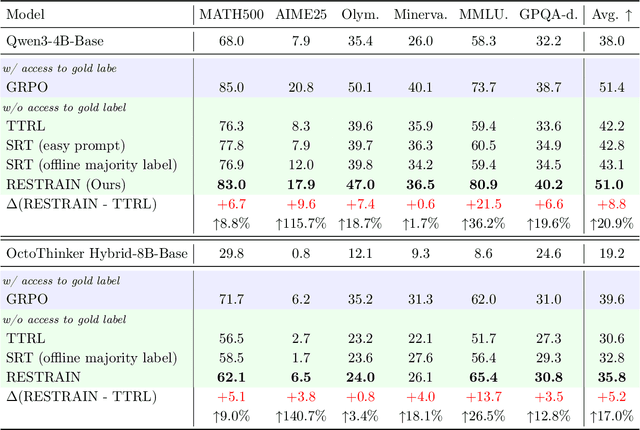
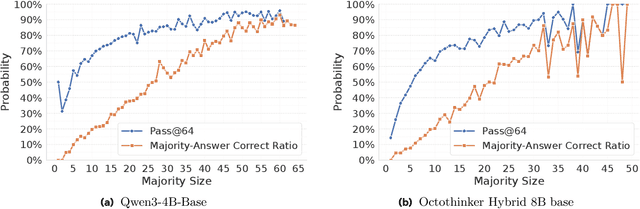
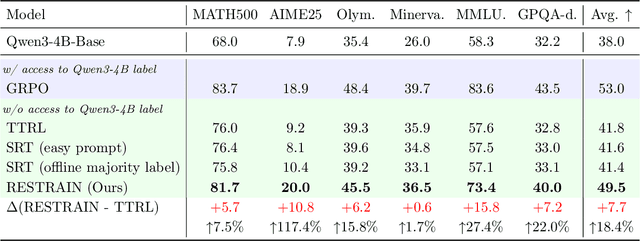
Reinforcement learning with human-annotated data has boosted chain-of-thought reasoning in large reasoning models, but these gains come at high costs in labeled data while faltering on harder tasks. A natural next step is experience-driven learning, where models improve without curated labels by adapting to unlabeled data. We introduce RESTRAIN (REinforcement learning with Self-restraint), a self-penalizing RL framework that converts the absence of gold labels into a useful learning signal. Instead of overcommitting to spurious majority votes, RESTRAIN exploits signals from the model's entire answer distribution: penalizing overconfident rollouts and low-consistency examples while preserving promising reasoning chains. The self-penalization mechanism integrates seamlessly into policy optimization methods such as GRPO, enabling continual self-improvement without supervision. On challenging reasoning benchmarks, RESTRAIN delivers large gains using only unlabeled data. With Qwen3-4B-Base and OctoThinker Hybrid-8B-Base, it improves Pass@1 by up to +140.7 percent on AIME25, +36.2 percent on MMLU_STEM, and +19.6 percent on GPQA-Diamond, nearly matching gold-label training while using no gold labels. These results demonstrate that RESTRAIN establishes a scalable path toward stronger reasoning without gold labels.
Global Convergence in Neural ODEs: Impact of Activation Functions
Sep 26, 2025Neural Ordinary Differential Equations (ODEs) have been successful in various applications due to their continuous nature and parameter-sharing efficiency. However, these unique characteristics also introduce challenges in training, particularly with respect to gradient computation accuracy and convergence analysis. In this paper, we address these challenges by investigating the impact of activation functions. We demonstrate that the properties of activation functions, specifically smoothness and nonlinearity, are critical to the training dynamics. Smooth activation functions guarantee globally unique solutions for both forward and backward ODEs, while sufficient nonlinearity is essential for maintaining the spectral properties of the Neural Tangent Kernel (NTK) during training. Together, these properties enable us to establish the global convergence of Neural ODEs under gradient descent in overparameterized regimes. Our theoretical findings are validated by numerical experiments, which not only support our analysis but also provide practical guidelines for scaling Neural ODEs, potentially leading to faster training and improved performance in real-world applications.
G2T-LLM: Graph-to-Tree Text Encoding for Molecule Generation with Fine-Tuned Large Language Models
Oct 03, 2024We introduce G2T-LLM, a novel approach for molecule generation that uses graph-to-tree text encoding to transform graph-based molecular structures into a hierarchical text format optimized for large language models (LLMs). This encoding converts complex molecular graphs into tree-structured formats, such as JSON and XML, which LLMs are particularly adept at processing due to their extensive pre-training on these types of data. By leveraging the flexibility of LLMs, our approach allows for intuitive interaction using natural language prompts, providing a more accessible interface for molecular design. Through supervised fine-tuning, G2T-LLM generates valid and coherent chemical structures, addressing common challenges like invalid outputs seen in traditional graph-based methods. While LLMs are computationally intensive, they offer superior generalization and adaptability, enabling the generation of diverse molecular structures with minimal task-specific customization. The proposed approach achieved comparable performances with state-of-the-art methods on various benchmark molecular generation datasets, demonstrating its potential as a flexible and innovative tool for AI-driven molecular design.
MAGE: Model-Level Graph Neural Networks Explanations via Motif-based Graph Generation
May 21, 2024Graph Neural Networks (GNNs) have shown remarkable success in molecular tasks, yet their interpretability remains challenging. Traditional model-level explanation methods like XGNN and GNNInterpreter often fail to identify valid substructures like rings, leading to questionable interpretability. This limitation stems from XGNN's atom-by-atom approach and GNNInterpreter's reliance on average graph embeddings, which overlook the essential structural elements crucial for molecules. To address these gaps, we introduce an innovative \textbf{M}otif-b\textbf{A}sed \textbf{G}NN \textbf{E}xplainer (MAGE) that uses motifs as fundamental units for generating explanations. Our approach begins with extracting potential motifs through a motif decomposition technique. Then, we utilize an attention-based learning method to identify class-specific motifs. Finally, we employ a motif-based graph generator for each class to create molecular graph explanations based on these class-specific motifs. This novel method not only incorporates critical substructures into the explanations but also guarantees their validity, yielding results that are human-understandable. Our proposed method's effectiveness is demonstrated through quantitative and qualitative assessments conducted on six real-world molecular datasets.
Inferring Data Preconditions from Deep Learning Models for Trustworthy Prediction in Deployment
Jan 26, 2024


Deep learning models are trained with certain assumptions about the data during the development stage and then used for prediction in the deployment stage. It is important to reason about the trustworthiness of the model's predictions with unseen data during deployment. Existing methods for specifying and verifying traditional software are insufficient for this task, as they cannot handle the complexity of DNN model architecture and expected outcomes. In this work, we propose a novel technique that uses rules derived from neural network computations to infer data preconditions for a DNN model to determine the trustworthiness of its predictions. Our approach, DeepInfer involves introducing a novel abstraction for a trained DNN model that enables weakest precondition reasoning using Dijkstra's Predicate Transformer Semantics. By deriving rules over the inductive type of neural network abstract representation, we can overcome the matrix dimensionality issues that arise from the backward non-linear computation from the output layer to the input layer. We utilize the weakest precondition computation using rules of each kind of activation function to compute layer-wise precondition from the given postcondition on the final output of a deep neural network. We extensively evaluated DeepInfer on 29 real-world DNN models using four different datasets collected from five different sources and demonstrated the utility, effectiveness, and performance improvement over closely related work. DeepInfer efficiently detects correct and incorrect predictions of high-accuracy models with high recall (0.98) and high F-1 score (0.84) and has significantly improved over prior technique, SelfChecker. The average runtime overhead of DeepInfer is low, 0.22 sec for all unseen datasets. We also compared runtime overhead using the same hardware settings and found that DeepInfer is 3.27 times faster than SelfChecker.
MotifPiece: A Data-Driven Approach for Effective Motif Extraction and Molecular Representation Learning
Dec 24, 2023Motif extraction is an important task in motif based molecular representation learning. Previously, machine learning approaches employing either rule-based or string-based techniques to extract motifs. Rule-based approaches may extract motifs that aren't frequent or prevalent within the molecular data, which can lead to an incomplete understanding of essential structural patterns in molecules. String-based methods often lose the topological information inherent in molecules. This can be a significant drawback because topology plays a vital role in defining the spatial arrangement and connectivity of atoms within a molecule, which can be critical for understanding its properties and behavior. In this paper, we develop a data-driven motif extraction technique known as MotifPiece, which employs statistical measures to define motifs. To comprehensively evaluate the effectiveness of MotifPiece, we introduce a heterogeneous learning module. Our model shows an improvement compared to previously reported models. Additionally, we demonstrate that its performance can be further enhanced in two ways: first, by incorporating more data to aid in generating a richer motif vocabulary, and second, by merging multiple datasets that share enough motifs, allowing for cross-dataset learning.
Wide Neural Networks as Gaussian Processes: Lessons from Deep Equilibrium Models
Oct 16, 2023
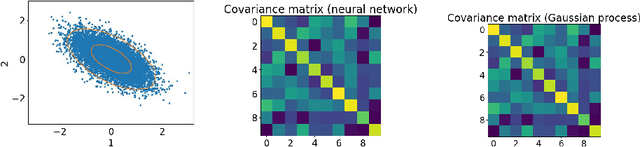
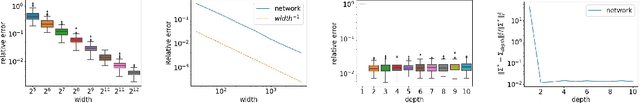
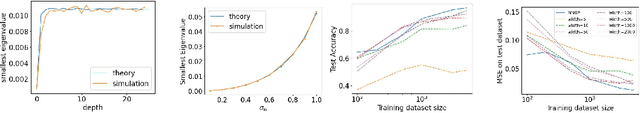
Neural networks with wide layers have attracted significant attention due to their equivalence to Gaussian processes, enabling perfect fitting of training data while maintaining generalization performance, known as benign overfitting. However, existing results mainly focus on shallow or finite-depth networks, necessitating a comprehensive analysis of wide neural networks with infinite-depth layers, such as neural ordinary differential equations (ODEs) and deep equilibrium models (DEQs). In this paper, we specifically investigate the deep equilibrium model (DEQ), an infinite-depth neural network with shared weight matrices across layers. Our analysis reveals that as the width of DEQ layers approaches infinity, it converges to a Gaussian process, establishing what is known as the Neural Network and Gaussian Process (NNGP) correspondence. Remarkably, this convergence holds even when the limits of depth and width are interchanged, which is not observed in typical infinite-depth Multilayer Perceptron (MLP) networks. Furthermore, we demonstrate that the associated Gaussian vector remains non-degenerate for any pairwise distinct input data, ensuring a strictly positive smallest eigenvalue of the corresponding kernel matrix using the NNGP kernel. These findings serve as fundamental elements for studying the training and generalization of DEQs, laying the groundwork for future research in this area.
DeepDFA: Dataflow Analysis-Guided Efficient Graph Learning for Vulnerability Detection
Dec 15, 2022



Deep learning-based vulnerability detection models have recently been shown to be effective and, in some cases, outperform static analysis tools. However, the highest-performing approaches use token-based transformer models, which do not leverage domain knowledge. Classical program analysis techniques such as dataflow analysis can detect many types of bugs and are the most commonly used methods in practice. Motivated by the causal relationship between bugs and dataflow analysis, we present DeepDFA, a dataflow analysis-guided graph learning framework and embedding that uses program semantic features for vulnerability detection. We show that DeepDFA is performant and efficient. DeepDFA ranked first in recall, first in generalizing over unseen projects, and second in F1 among all the state-of-the-art models we experimented with. It is also the smallest model in terms of the number of parameters, and was trained in 9 minutes, 69x faster than the highest-performing baseline. DeepDFA can be used with other models. By integrating LineVul and DeepDFA, we achieved the best vulnerability detection performance of 96.4 F1 score, 98.69 precision, and 94.22 recall.
On the optimization and generalization of overparameterized implicit neural networks
Sep 30, 2022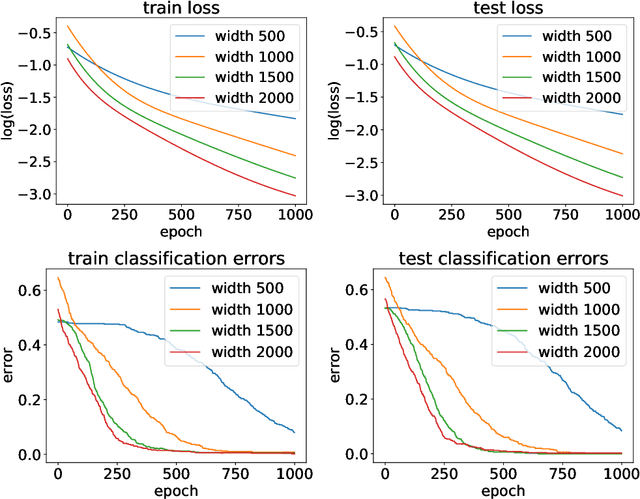
Implicit neural networks have become increasingly attractive in the machine learning community since they can achieve competitive performance but use much less computational resources. Recently, a line of theoretical works established the global convergences for first-order methods such as gradient descent if the implicit networks are over-parameterized. However, as they train all layers together, their analyses are equivalent to only studying the evolution of the output layer. It is unclear how the implicit layer contributes to the training. Thus, in this paper, we restrict ourselves to only training the implicit layer. We show that global convergence is guaranteed, even if only the implicit layer is trained. On the other hand, the theoretical understanding of when and how the training performance of an implicit neural network can be generalized to unseen data is still under-explored. Although this problem has been studied in standard feed-forward networks, the case of implicit neural networks is still intriguing since implicit networks theoretically have infinitely many layers. Therefore, this paper investigates the generalization error for implicit neural networks. Specifically, we study the generalization of an implicit network activated by the ReLU function over random initialization. We provide a generalization bound that is initialization sensitive. As a result, we show that gradient flow with proper random initialization can train a sufficient over-parameterized implicit network to achieve arbitrarily small generalization errors.