 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRESTRAIN: From Spurious Votes to Signals -- Self-Driven RL with Self-Penalization
Oct 02, 2025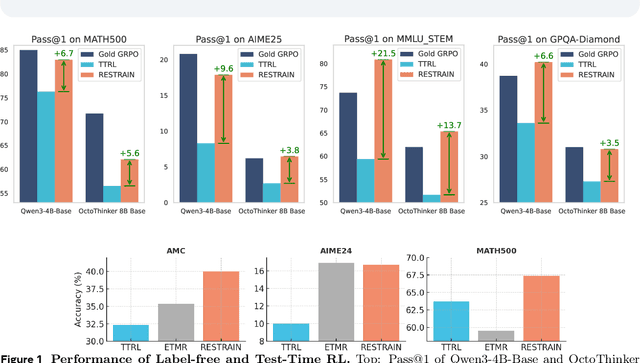
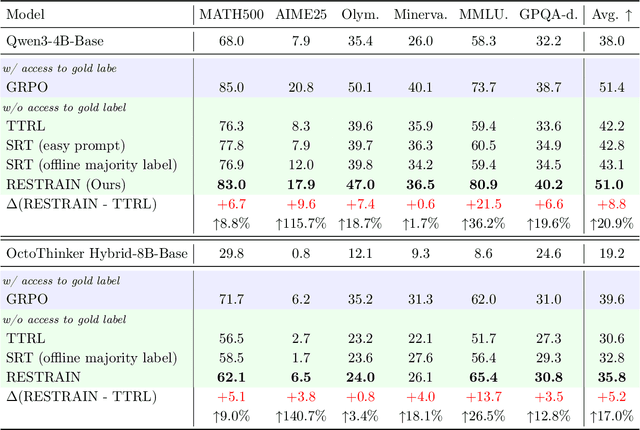
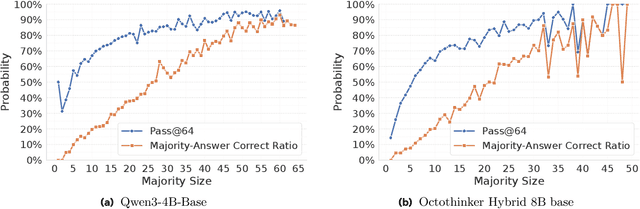
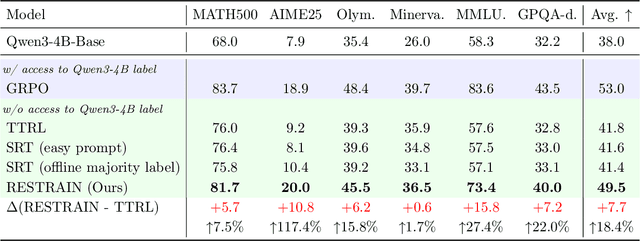
Reinforcement learning with human-annotated data has boosted chain-of-thought reasoning in large reasoning models, but these gains come at high costs in labeled data while faltering on harder tasks. A natural next step is experience-driven learning, where models improve without curated labels by adapting to unlabeled data. We introduce RESTRAIN (REinforcement learning with Self-restraint), a self-penalizing RL framework that converts the absence of gold labels into a useful learning signal. Instead of overcommitting to spurious majority votes, RESTRAIN exploits signals from the model's entire answer distribution: penalizing overconfident rollouts and low-consistency examples while preserving promising reasoning chains. The self-penalization mechanism integrates seamlessly into policy optimization methods such as GRPO, enabling continual self-improvement without supervision. On challenging reasoning benchmarks, RESTRAIN delivers large gains using only unlabeled data. With Qwen3-4B-Base and OctoThinker Hybrid-8B-Base, it improves Pass@1 by up to +140.7 percent on AIME25, +36.2 percent on MMLU_STEM, and +19.6 percent on GPQA-Diamond, nearly matching gold-label training while using no gold labels. These results demonstrate that RESTRAIN establishes a scalable path toward stronger reasoning without gold labels.
G2T-LLM: Graph-to-Tree Text Encoding for Molecule Generation with Fine-Tuned Large Language Models
Oct 03, 2024We introduce G2T-LLM, a novel approach for molecule generation that uses graph-to-tree text encoding to transform graph-based molecular structures into a hierarchical text format optimized for large language models (LLMs). This encoding converts complex molecular graphs into tree-structured formats, such as JSON and XML, which LLMs are particularly adept at processing due to their extensive pre-training on these types of data. By leveraging the flexibility of LLMs, our approach allows for intuitive interaction using natural language prompts, providing a more accessible interface for molecular design. Through supervised fine-tuning, G2T-LLM generates valid and coherent chemical structures, addressing common challenges like invalid outputs seen in traditional graph-based methods. While LLMs are computationally intensive, they offer superior generalization and adaptability, enabling the generation of diverse molecular structures with minimal task-specific customization. The proposed approach achieved comparable performances with state-of-the-art methods on various benchmark molecular generation datasets, demonstrating its potential as a flexible and innovative tool for AI-driven molecular design.
MAGE: Model-Level Graph Neural Networks Explanations via Motif-based Graph Generation
May 21, 2024Graph Neural Networks (GNNs) have shown remarkable success in molecular tasks, yet their interpretability remains challenging. Traditional model-level explanation methods like XGNN and GNNInterpreter often fail to identify valid substructures like rings, leading to questionable interpretability. This limitation stems from XGNN's atom-by-atom approach and GNNInterpreter's reliance on average graph embeddings, which overlook the essential structural elements crucial for molecules. To address these gaps, we introduce an innovative \textbf{M}otif-b\textbf{A}sed \textbf{G}NN \textbf{E}xplainer (MAGE) that uses motifs as fundamental units for generating explanations. Our approach begins with extracting potential motifs through a motif decomposition technique. Then, we utilize an attention-based learning method to identify class-specific motifs. Finally, we employ a motif-based graph generator for each class to create molecular graph explanations based on these class-specific motifs. This novel method not only incorporates critical substructures into the explanations but also guarantees their validity, yielding results that are human-understandable. Our proposed method's effectiveness is demonstrated through quantitative and qualitative assessments conducted on six real-world molecular datasets.
MotifPiece: A Data-Driven Approach for Effective Motif Extraction and Molecular Representation Learning
Dec 24, 2023Motif extraction is an important task in motif based molecular representation learning. Previously, machine learning approaches employing either rule-based or string-based techniques to extract motifs. Rule-based approaches may extract motifs that aren't frequent or prevalent within the molecular data, which can lead to an incomplete understanding of essential structural patterns in molecules. String-based methods often lose the topological information inherent in molecules. This can be a significant drawback because topology plays a vital role in defining the spatial arrangement and connectivity of atoms within a molecule, which can be critical for understanding its properties and behavior. In this paper, we develop a data-driven motif extraction technique known as MotifPiece, which employs statistical measures to define motifs. To comprehensively evaluate the effectiveness of MotifPiece, we introduce a heterogeneous learning module. Our model shows an improvement compared to previously reported models. Additionally, we demonstrate that its performance can be further enhanced in two ways: first, by incorporating more data to aid in generating a richer motif vocabulary, and second, by merging multiple datasets that share enough motifs, allowing for cross-dataset learning.
Molecular Graph Representation Learning via Heterogeneous Motif Graph Construction
Feb 01, 2022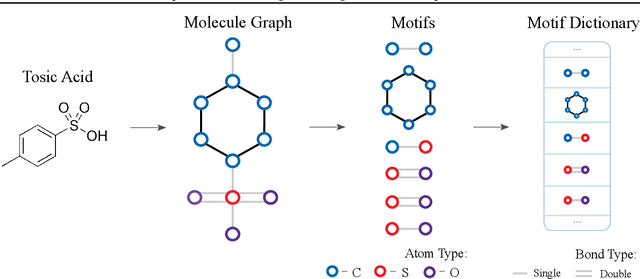
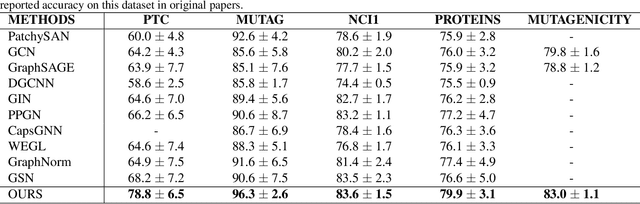
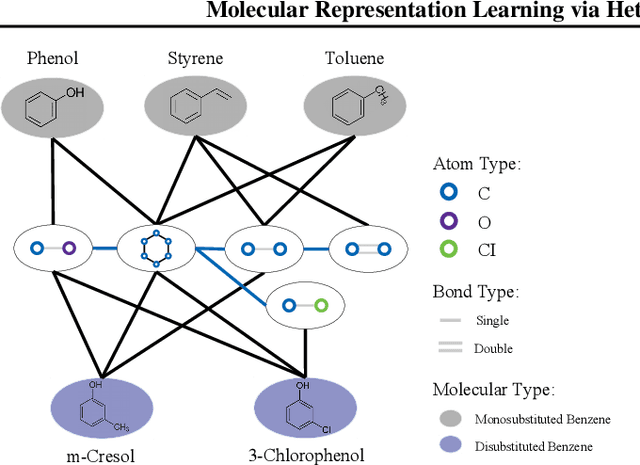
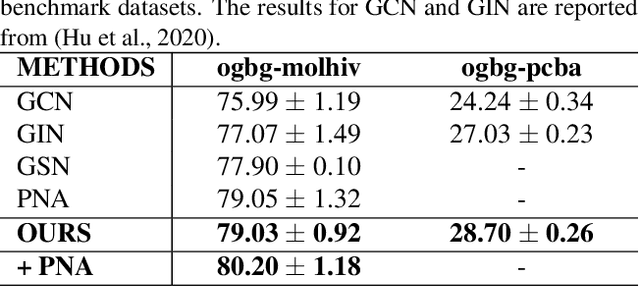
We consider feature representation learning problem of molecular graphs. Graph Neural Networks have been widely used in feature representation learning of molecular graphs. However, most existing methods deal with molecular graphs individually while neglecting their connections, such as motif-level relationships. We propose a novel molecular graph representation learning method by constructing a heterogeneous motif graph to address this issue. In particular, we build a heterogeneous motif graph that contains motif nodes and molecular nodes. Each motif node corresponds to a motif extracted from molecules. Then, we propose a Heterogeneous Motif Graph Neural Network (HM-GNN) to learn feature representations for each node in the heterogeneous motif graph. Our heterogeneous motif graph also enables effective multi-task learning, especially for small molecular datasets. To address the potential efficiency issue, we propose to use an edge sampler, which can significantly reduce computational resources usage. The experimental results show that our model consistently outperforms previous state-of-the-art models. Under multi-task settings, the promising performances of our methods on combined datasets shed light on a new learning paradigm for small molecular datasets. Finally, we show that our model achieves similar performances with significantly less computational resources by using our edge sampler.
MotifExplainer: a Motif-based Graph Neural Network Explainer
Feb 01, 2022We consider the explanation problem of Graph Neural Networks (GNNs). Most existing GNN explanation methods identify the most important edges or nodes but fail to consider substructures, which are more important for graph data. The only method that considers subgraphs tries to search all possible subgraphs and identify the most significant subgraphs. However, the subgraphs identified may not be recurrent or statistically important. In this work, we propose a novel method, known as MotifExplainer, to explain GNNs by identifying important motifs, recurrent and statistically significant patterns in graphs. Our proposed motif-based methods can provide better human-understandable explanations than methods based on nodes, edges, and regular subgraphs. Given an input graph and a pre-trained GNN model, our method first extracts motifs in the graph using well-designed motif extraction rules. Then we generate motif embedding by feeding motifs into the pre-trained GNN. Finally, we employ an attention-based method to identify the most influential motifs as explanations for the final prediction results. The empirical studies on both synthetic and real-world datasets demonstrate the effectiveness of our method.