 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeData Transfer Approaches to Improve Seq-to-Seq Retrosynthesis
Oct 02, 2020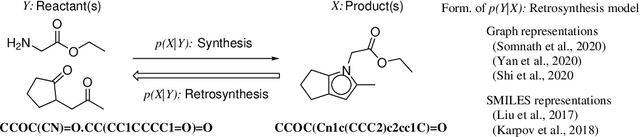

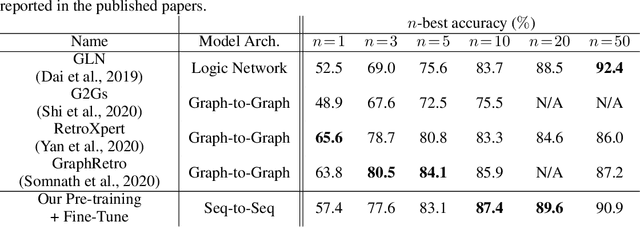
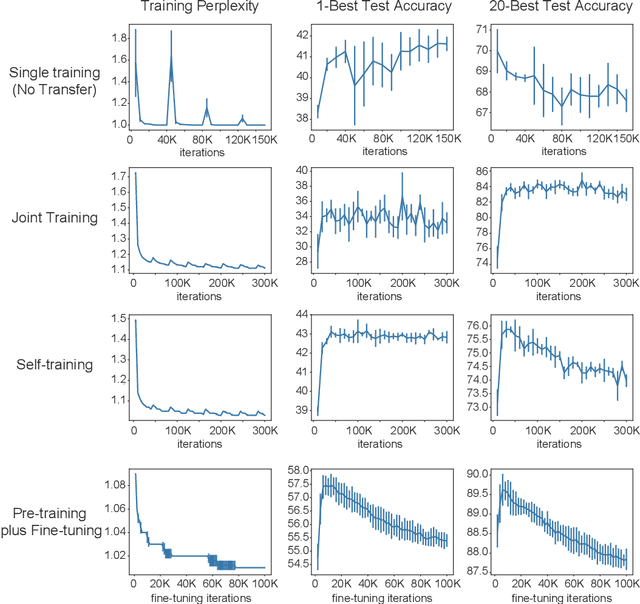
Retrosynthesis is a problem to infer reactant compounds to synthesize a given product compound through chemical reactions. Recent studies on retrosynthesis focus on proposing more sophisticated prediction models, but the dataset to feed the models also plays an essential role in achieving the best generalizing models. Generally, a dataset that is best suited for a specific task tends to be small. In such a case, it is the standard solution to transfer knowledge from a large or clean dataset in the same domain. In this paper, we conduct a systematic and intensive examination of data transfer approaches on end-to-end generative models, in application to retrosynthesis. Experimental results show that typical data transfer methods can improve test prediction scores of an off-the-shelf Transformer baseline model. Especially, the pre-training plus fine-tuning approach boosts the accuracy scores of the baseline, achieving the new state-of-the-art. In addition, we conduct a manual inspection for the erroneous prediction results. The inspection shows that the pre-training plus fine-tuning models can generate chemically appropriate or sensible proposals in almost all cases.
Graph Residual Flow for Molecular Graph Generation
Sep 30, 2019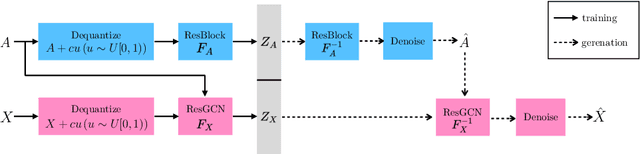
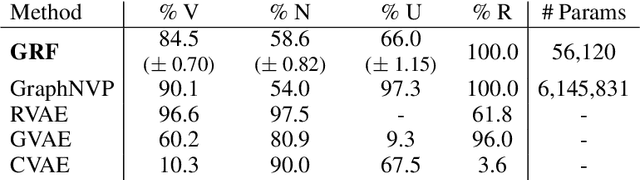
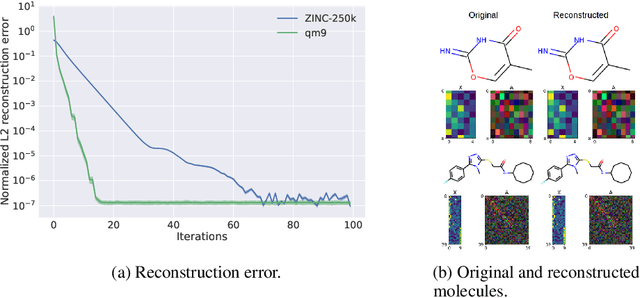
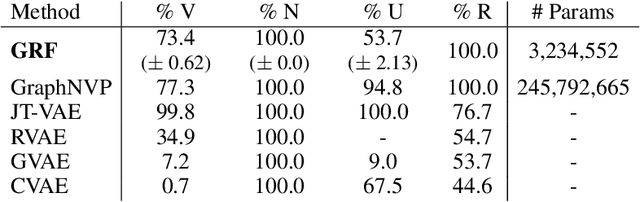
Statistical generative models for molecular graphs attract attention from many researchers from the fields of bio- and chemo-informatics. Among these models, invertible flow-based approaches are not fully explored yet. In this paper, we propose a powerful invertible flow for molecular graphs, called graph residual flow (GRF). The GRF is based on residual flows, which are known for more flexible and complex non-linear mappings than traditional coupling flows. We theoretically derive non-trivial conditions such that GRF is invertible, and present a way of keeping the entire flows invertible throughout the training and sampling. Experimental results show that a generative model based on the proposed GRF achieves comparable generation performance, with much smaller number of trainable parameters compared to the existing flow-based model.
BayesGrad: Explaining Predictions of Graph Convolutional Networks
Jul 04, 2018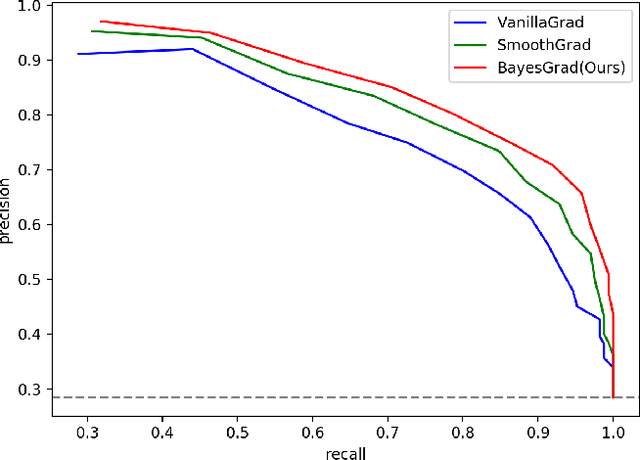
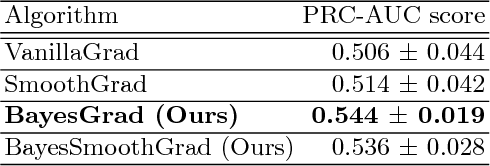
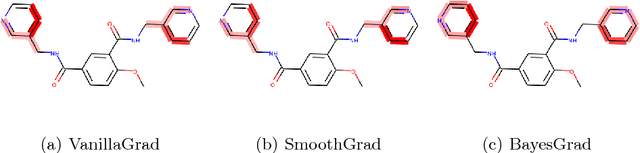
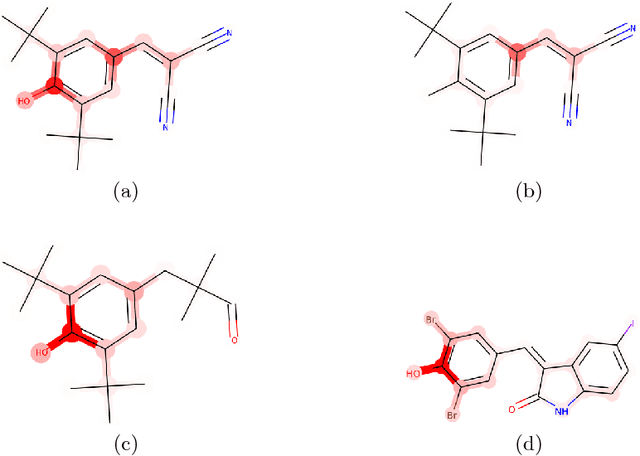
Recent advances in graph convolutional networks have significantly improved the performance of chemical predictions, raising a new research question: "how do we explain the predictions of graph convolutional networks?" A possible approach to answer this question is to visualize evidence substructures responsible for the predictions. For chemical property prediction tasks, the sample size of the training data is often small and/or a label imbalance problem occurs, where a few samples belong to a single class and the majority of samples belong to the other classes. This can lead to uncertainty related to the learned parameters of the machine learning model. To address this uncertainty, we propose BayesGrad, utilizing the Bayesian predictive distribution, to define the importance of each node in an input graph, which is computed efficiently using the dropout technique. We demonstrate that BayesGrad successfully visualizes the substructures responsible for the label prediction in the artificial experiment, even when the sample size is small. Furthermore, we use a real dataset to evaluate the effectiveness of the visualization. The basic idea of BayesGrad is not limited to graph-structured data and can be applied to other data types.