 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDISHONEST: Dissecting misInformation Spread using Homogeneous sOcial NEtworks and Semantic Topic classification
Dec 12, 2024The emergence of the COVID-19 pandemic resulted in a significant rise in the spread of misinformation on online platforms such as Twitter. Oftentimes this growth is blamed on the idea of the "echo chamber." However, the behavior said to characterize these echo chambers exists in two dimensions. The first is in a user's social interactions, where they are said to stick with the same clique of like-minded users. The second is in the content of their posts, where they are said to repeatedly espouse homogeneous ideas. In this study, we link the two by using Twitter's network of retweets to study social interactions and topic modeling to study tweet content. In order to measure the diversity of a user's interactions over time, we develop a novel metric to track the speed at which they travel through the social network. The application of these analysis methods to misinformation-focused data from the pandemic demonstrates correlation between social behavior and tweet content. We believe this correlation supports the common intuition about how antisocial users behave, and further suggests that it holds even in subcommunities already rife with misinformation.
Unsupervised Keyphrase Extraction via Interpretable Neural Networks
Mar 15, 2022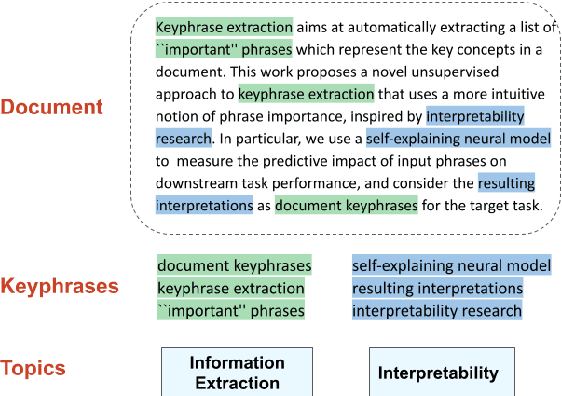
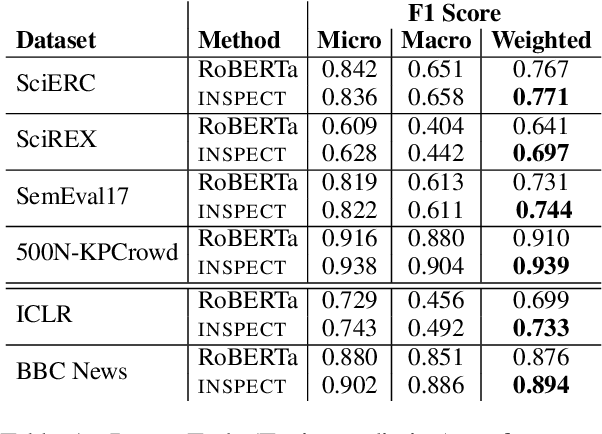
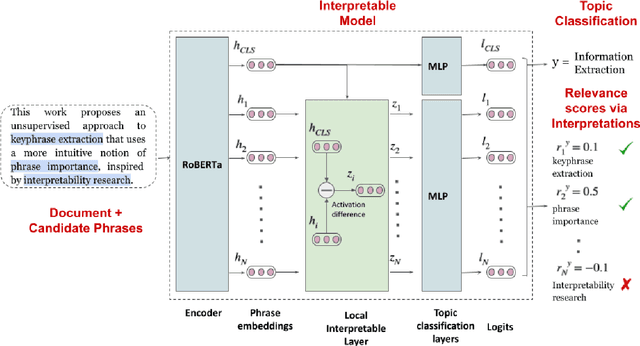
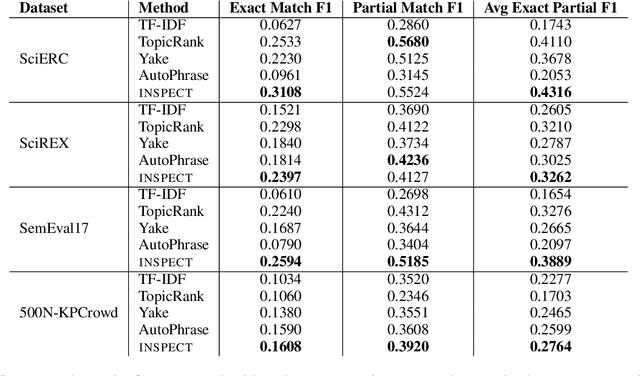
Keyphrase extraction aims at automatically extracting a list of "important" phrases which represent the key concepts in a document. Prior approaches for unsupervised keyphrase extraction resort to heuristic notions of phrase importance via embedding similarities or graph centrality, requiring extensive domain expertise to develop them. Our work proposes an alternative operational definition: phrases that are most useful for predicting the topic of a text are important keyphrases. To this end, we propose INSPECT -- a self-explaining neural framework for identifying influential keyphrases by measuring the predictive impact of input phrases on the downstream task of topic classification. We show that this novel approach not only alleviates the need for ad-hoc heuristics but also achieves state-of-the-art results in unsupervised keyphrase extraction across four diverse datasets in two domains: scientific publications and news articles. Ultimately, our study suggests a new usage of interpretable neural networks as an intrinsic component in NLP systems, and not only as a tool for explaining model predictions to humans.
Predicting Aqueous Solubility of Organic Molecules Using Deep Learning Models with Varied Molecular Representations
May 27, 2021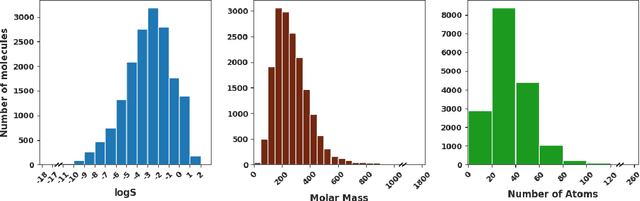
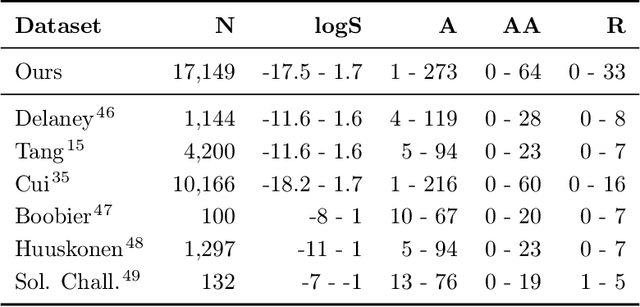
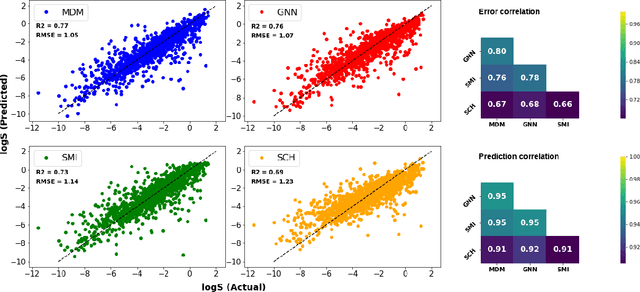
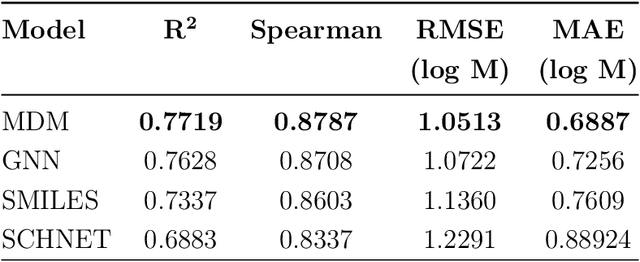
Determining the aqueous solubility of molecules is a vital step in many pharmaceutical, environmental, and energy storage applications. Despite efforts made over decades, there are still challenges associated with developing a solubility prediction model with satisfactory accuracy for many of these applications. The goal of this study is to develop a general model capable of predicting the solubility of a broad range of organic molecules. Using the largest currently available solubility dataset, we implement deep learning-based models to predict solubility from molecular structure and explore several different molecular representations including molecular descriptors, simplified molecular-input line-entry system (SMILES) strings, molecular graphs, and three-dimensional (3D) atomic coordinates using four different neural network architectures - fully connected neural networks (FCNNs), recurrent neural networks (RNNs), graph neural networks (GNNs), and SchNet. We find that models using molecular descriptors achieve the best performance, with GNN models also achieving good performance. We perform extensive error analysis to understand the molecular properties that influence model performance, perform feature analysis to understand which information about molecular structure is most valuable for prediction, and perform a transfer learning and data size study to understand the impact of data availability on model performance.