 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraphical Gaussian Process Regression Model for Aqueous Solvation Free Energy Prediction of Organic Molecules in Redox Flow Battery
Jun 15, 2021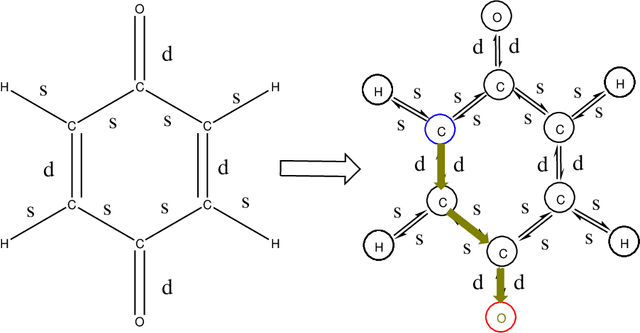
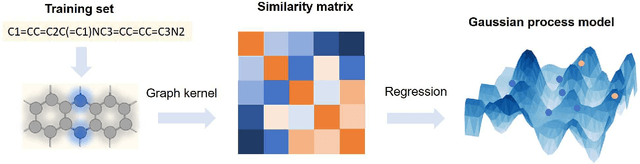
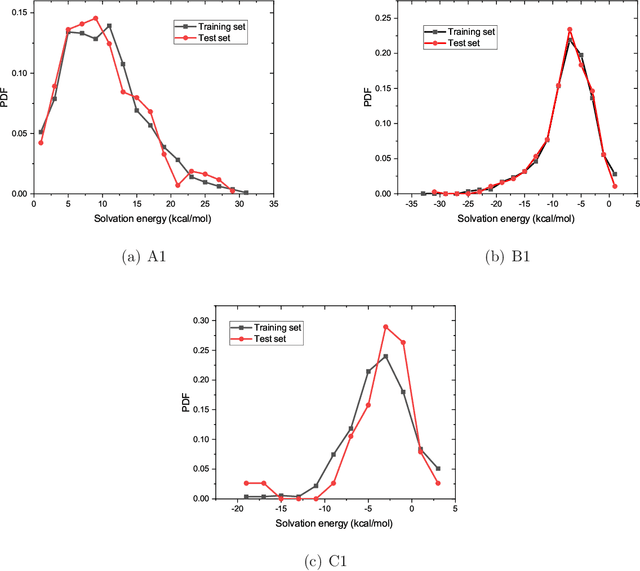
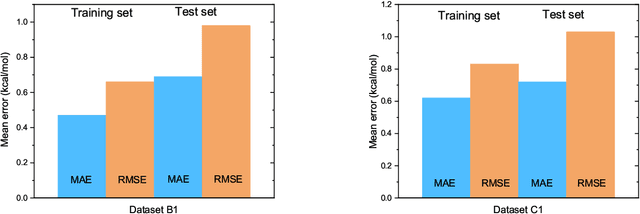
The solvation free energy of organic molecules is a critical parameter in determining emergent properties such as solubility, liquid-phase equilibrium constants, and pKa and redox potentials in an organic redox flow battery. In this work, we present a machine learning (ML) model that can learn and predict the aqueous solvation free energy of an organic molecule using Gaussian process regression method based on a new molecular graph kernel. To investigate the performance of the ML model on electrostatic interaction, the nonpolar interaction contribution of solvent and the conformational entropy of solute in solvation free energy, three data sets with implicit or explicit water solvent models, and contribution of conformational entropy of solute are tested. We demonstrate that our ML model can predict the solvation free energy of molecules at chemical accuracy with a mean absolute error of less than 1 kcal/mol for subsets of the QM9 dataset and the Freesolv database. To solve the general data scarcity problem for a graph-based ML model, we propose a dimension reduction algorithm based on the distance between molecular graphs, which can be used to examine the diversity of the molecular data set. It provides a promising way to build a minimum training set to improve prediction for certain test sets where the space of molecular structures is predetermined.
Predicting Aqueous Solubility of Organic Molecules Using Deep Learning Models with Varied Molecular Representations
May 27, 2021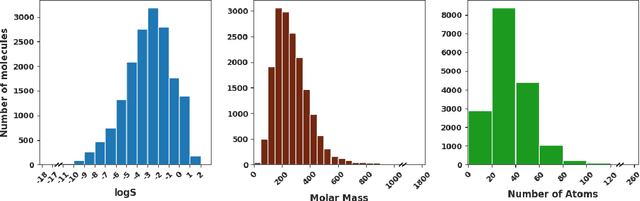
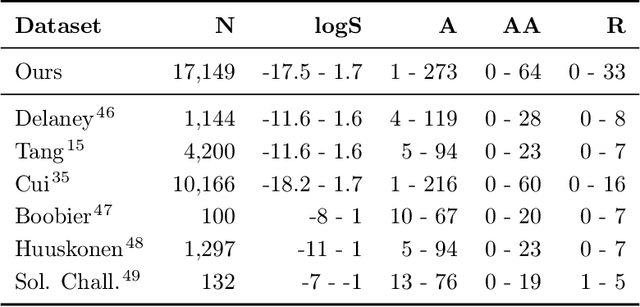
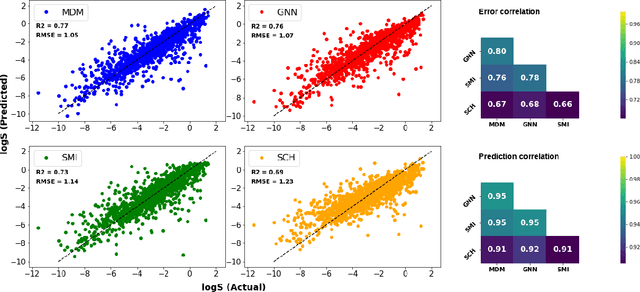
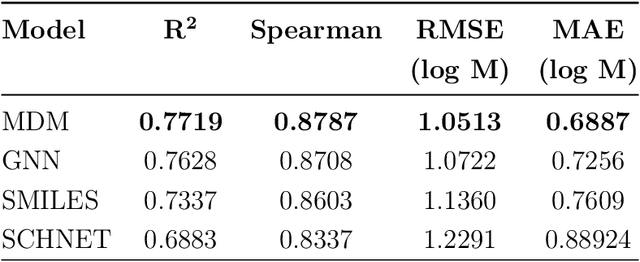
Determining the aqueous solubility of molecules is a vital step in many pharmaceutical, environmental, and energy storage applications. Despite efforts made over decades, there are still challenges associated with developing a solubility prediction model with satisfactory accuracy for many of these applications. The goal of this study is to develop a general model capable of predicting the solubility of a broad range of organic molecules. Using the largest currently available solubility dataset, we implement deep learning-based models to predict solubility from molecular structure and explore several different molecular representations including molecular descriptors, simplified molecular-input line-entry system (SMILES) strings, molecular graphs, and three-dimensional (3D) atomic coordinates using four different neural network architectures - fully connected neural networks (FCNNs), recurrent neural networks (RNNs), graph neural networks (GNNs), and SchNet. We find that models using molecular descriptors achieve the best performance, with GNN models also achieving good performance. We perform extensive error analysis to understand the molecular properties that influence model performance, perform feature analysis to understand which information about molecular structure is most valuable for prediction, and perform a transfer learning and data size study to understand the impact of data availability on model performance.