 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrediction of Hereditary Cancers Using Neural Networks
Jun 25, 2021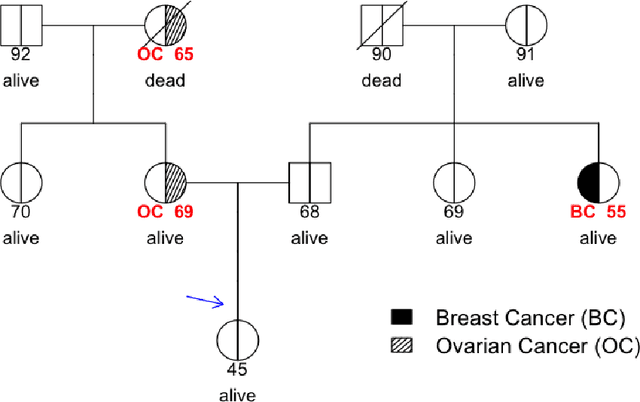
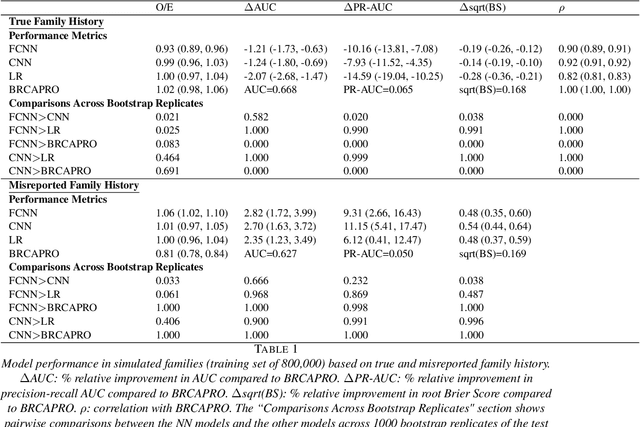
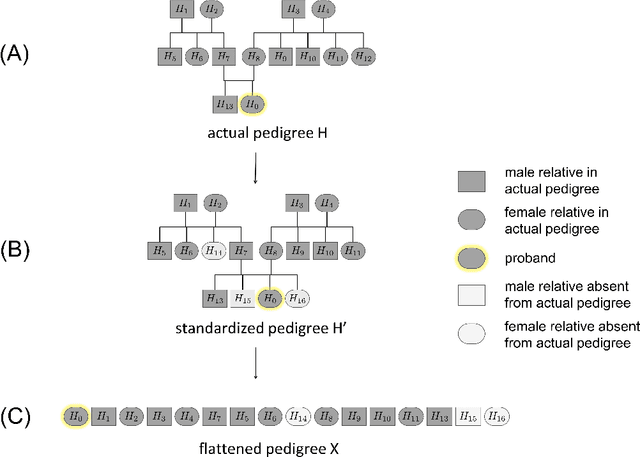
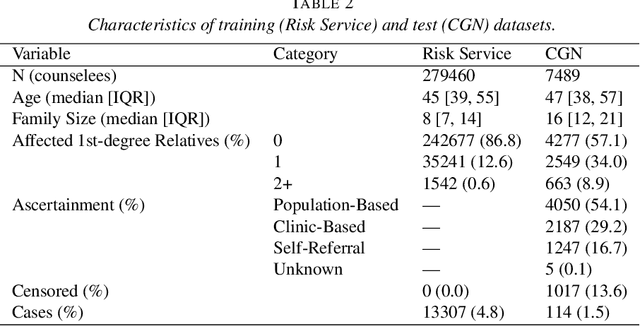
Family history is a major risk factor for many types of cancer. Mendelian risk prediction models translate family histories into cancer risk predictions based on knowledge of cancer susceptibility genes. These models are widely used in clinical practice to help identify high-risk individuals. Mendelian models leverage the entire family history, but they rely on many assumptions about cancer susceptibility genes that are either unrealistic or challenging to validate due to low mutation prevalence. Training more flexible models, such as neural networks, on large databases of pedigrees can potentially lead to accuracy gains. In this paper, we develop a framework to apply neural networks to family history data and investigate their ability to learn inherited susceptibility to cancer. While there is an extensive literature on neural networks and their state-of-the-art performance in many tasks, there is little work applying them to family history data. We propose adaptations of fully-connected neural networks and convolutional neural networks to pedigrees. In data simulated under Mendelian inheritance, we demonstrate that our proposed neural network models are able to achieve nearly optimal prediction performance. Moreover, when the observed family history includes misreported cancer diagnoses, neural networks are able to outperform the Mendelian BRCAPRO model embedding the correct inheritance laws. Using a large dataset of over 200,000 family histories, the Risk Service cohort, we train prediction models for future risk of breast cancer. We validate the models using data from the Cancer Genetics Network.
Extending Models Via Gradient Boosting: An Application to Mendelian Models
May 13, 2021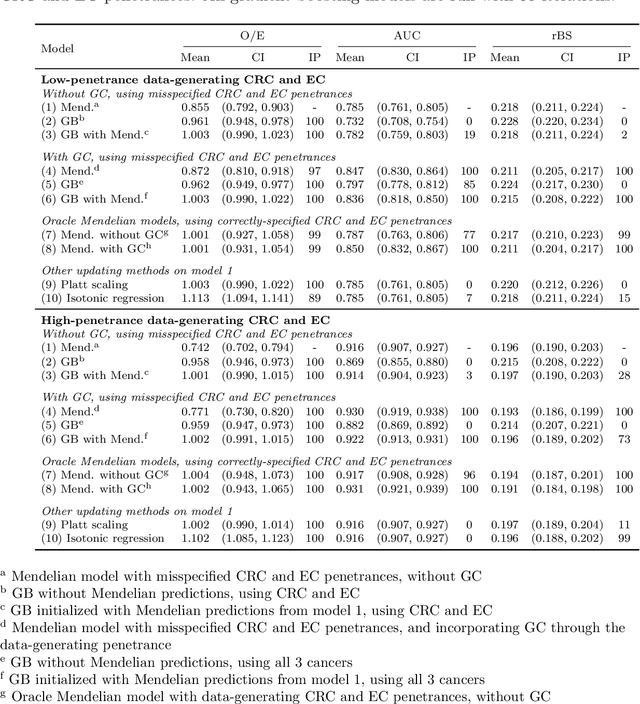
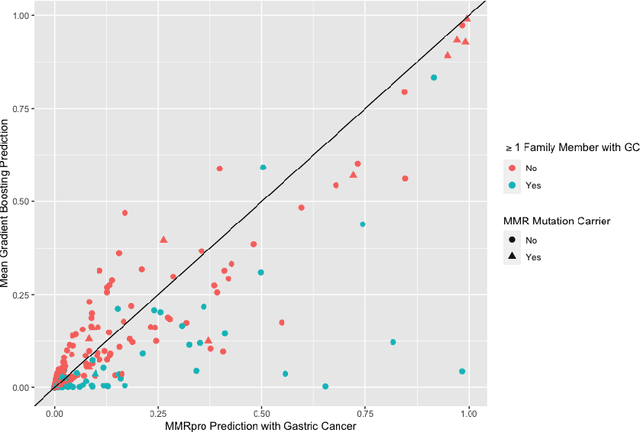
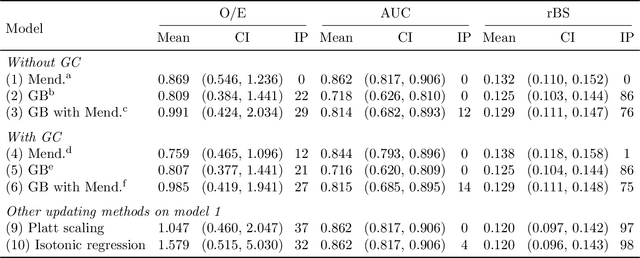
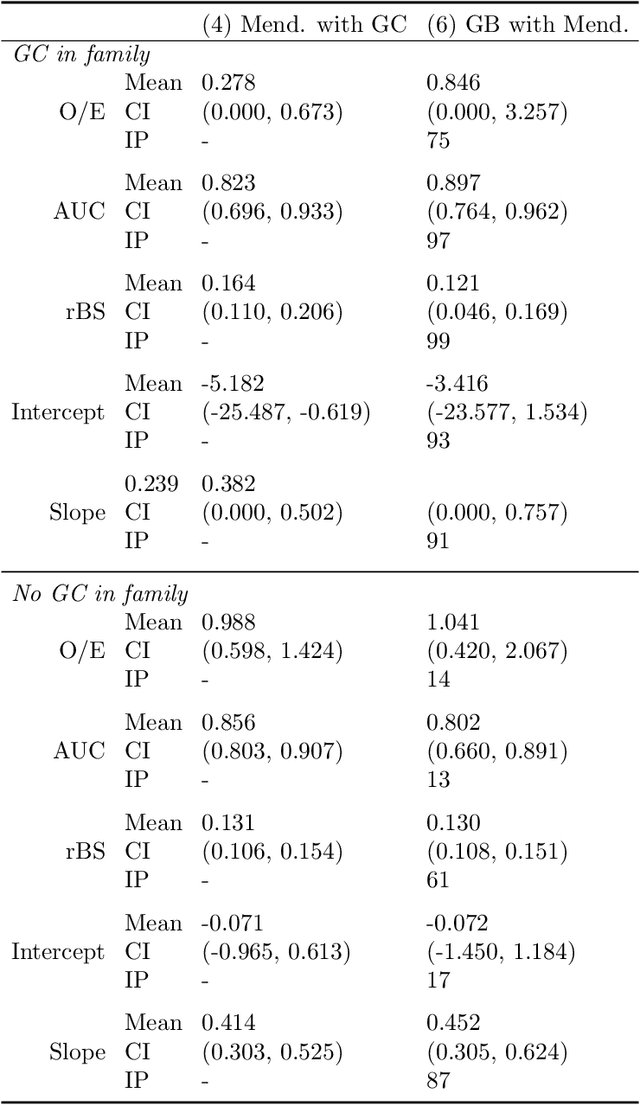
Improving existing widely-adopted prediction models is often a more efficient and robust way towards progress than training new models from scratch. Existing models may (a) incorporate complex mechanistic knowledge, (b) leverage proprietary information and, (c) have surmounted barriers to adoption. Compared to model training, model improvement and modification receive little attention. In this paper we propose a general approach to model improvement: we combine gradient boosting with any previously developed model to improve model performance while retaining important existing characteristics. To exemplify, we consider the context of Mendelian models, which estimate the probability of carrying genetic mutations that confer susceptibility to disease by using family pedigrees and health histories of family members. Via simulations we show that integration of gradient boosting with an existing Mendelian model can produce an improved model that outperforms both that model and the model built using gradient boosting alone. We illustrate the approach on genetic testing data from the USC-Stanford Cancer Genetics Hereditary Cancer Panel (HCP) study.
Using Machine Learning and Natural Language Processing to Review and Classify the Medical Literature on Cancer Susceptibility Genes
Apr 24, 2019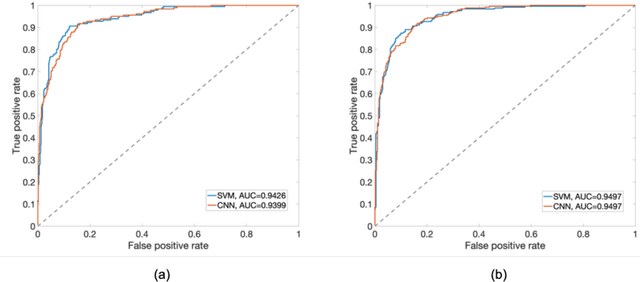
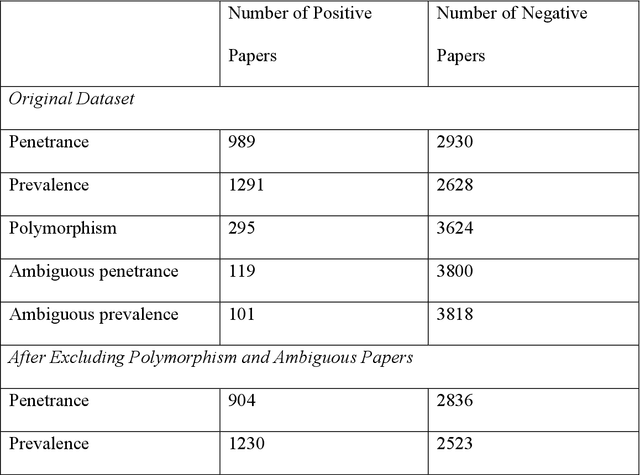
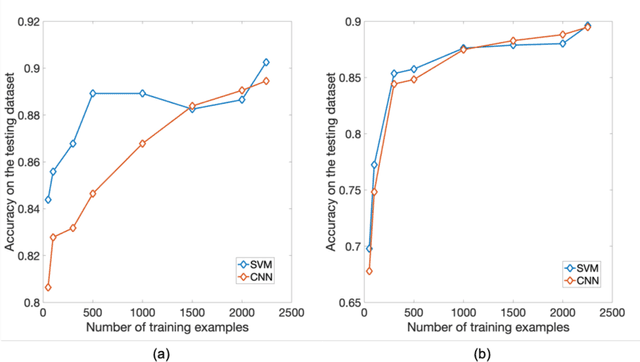
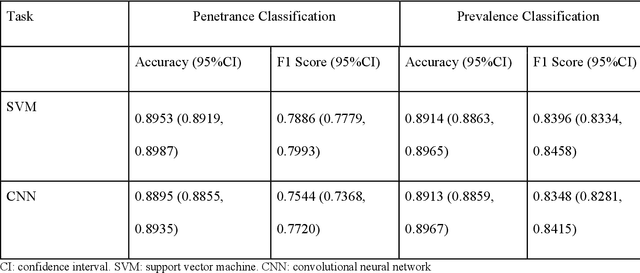
PURPOSE: The medical literature relevant to germline genetics is growing exponentially. Clinicians need tools monitoring and prioritizing the literature to understand the clinical implications of the pathogenic genetic variants. We developed and evaluated two machine learning models to classify abstracts as relevant to the penetrance (risk of cancer for germline mutation carriers) or prevalence of germline genetic mutations. METHODS: We conducted literature searches in PubMed and retrieved paper titles and abstracts to create an annotated dataset for training and evaluating the two machine learning classification models. Our first model is a support vector machine (SVM) which learns a linear decision rule based on the bag-of-ngrams representation of each title and abstract. Our second model is a convolutional neural network (CNN) which learns a complex nonlinear decision rule based on the raw title and abstract. We evaluated the performance of the two models on the classification of papers as relevant to penetrance or prevalence. RESULTS: For penetrance classification, we annotated 3740 paper titles and abstracts and used 60% for training the model, 20% for tuning the model, and 20% for evaluating the model. The SVM model achieves 89.53% accuracy (percentage of papers that were correctly classified) while the CNN model achieves 88.95 % accuracy. For prevalence classification, we annotated 3753 paper titles and abstracts. The SVM model achieves 89.14% accuracy while the CNN model achieves 89.13 % accuracy. CONCLUSION: Our models achieve high accuracy in classifying abstracts as relevant to penetrance or prevalence. By facilitating literature review, this tool could help clinicians and researchers keep abreast of the burgeoning knowledge of gene-cancer associations and keep the knowledge bases for clinical decision support tools up to date.
airpred: A Flexible R Package Implementing Methods for Predicting Air Pollution
Oct 30, 2018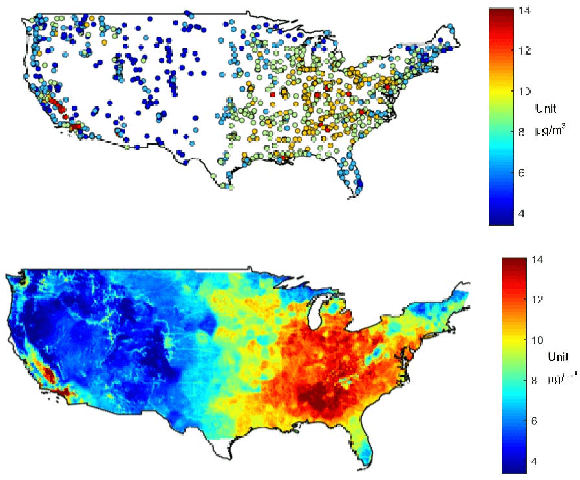

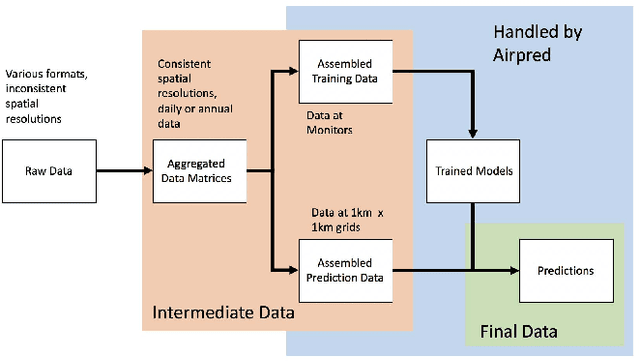
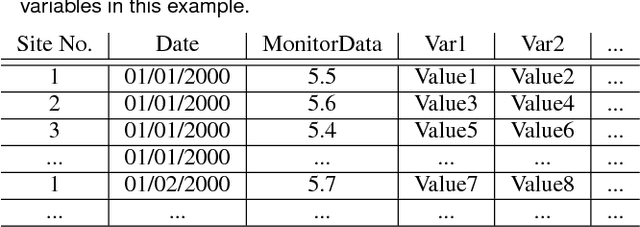
Fine particulate matter (PM$_{2.5}$) is one of the criteria air pollutants regulated by the Environmental Protection Agency in the United States. There is strong evidence that ambient exposure to (PM$_{2.5}$) increases risk of mortality and hospitalization. Large scale epidemiological studies on the health effects of PM$_{2.5}$ provide the necessary evidence base for lowering the safety standards and inform regulatory policy. However, ambient monitors of PM$_{2.5}$ (as well as monitors for other pollutants) are sparsely located across the U.S., and therefore studies based only on the levels of PM$_{2.5}$ measured from the monitors would inevitably exclude large amounts of the population. One approach to resolving this issue has been developing models to predict local PM$_{2.5}$, NO$_2$, and ozone based on satellite, meteorological, and land use data. This process typically relies developing a prediction model that relies on large amounts of input data and is highly computationally intensive to predict levels of air pollution in unmonitored areas. We have developed a flexible R package that allows for environmental health researchers to design and train spatio-temporal models capable of predicting multiple pollutants, including PM$_{2.5}$. We utilize H2O, an open source big data platform, to achieve both performance and scalability when used in conjunction with cloud or cluster computing systems.